High-pressure tubes
Posted by metzstein, on 6 April 2016
Notes on “Intracellular lumen formation in Drosophila proceeds via a novel subcellular compartment” by Linda S. Nikolova and Mark M. Metzstein. Development 2015 142: 3964-3973; doi: 10.1242/dev.127902
In this post, I provide additional details to a paper which we published last year in Development. In particular, I expand on our description on the method of high pressure freezing/freeze substitution, as well as why we developed this technique to examine Drosophila tracheal terminal cells, a part of the insect breathing system.
Like all terrestrial animals, insects require the ability to take oxygen from the atmosphere and deliver it to internal tissues. In vertebrates, this function is carried out by two independent systems: the lungs, used to take air into the body, and the vasculature, used to distribute oxygen throughout the body. Insects and other invertebrates use a different strategy, in which gas intake is directly coupled to the distribution system. This organization is accomplished by a single network of epithelial tubes, called trachea by analogy with vertebrate breathing systems. Openings on the surface of the insect, called spiracles, connect to large, multicellular tracheal tubes. In turn, smaller, unicellular tubes branch off the multicellular tubes and extend toward different regions of the animal. Finally, located at the ends of unicellular tubes, a number of specialized cells, known as terminal cells, are responsible for distribution of gas to all individual cells and tissues. Insect respiration is thought to be driven entirely by passive diffusion of air through the tracheal network, a method of respiration that obviously works well for this class of animals given their huge numbers and species diversity. However, it is also likely this passive diffusion of air sets the limits to organismal size, thus explaining why giant ants do not regularly rampage over the countryside.
We are particularly interested in the cell biological mechanisms of terminal cell morphogenesis. To perform their function, terminal cells undergo fascinating cell shape changes, with each individual cell undergoing an iterative process of plasma membrane outgrowth and bifurcation during larval stages. Eventually, each cell produces 20-100 thin (typically less than 1µm in diameter) subcellular branches (labeled with a cytoplasmic GFP in Fig. 1A). In terms of complexity, terminal cells rival the elaborate axonal and dendritic arbors found in neurons. However, unlike neurons, terminal cells have to undergo an additional form of morphogenesis: tubulogenesis. For gas to flow efficiently, each of the thin branches terminal cell branches develops into a tube, with a membrane-bound, gas-filled open space, called a lumen, running through it.
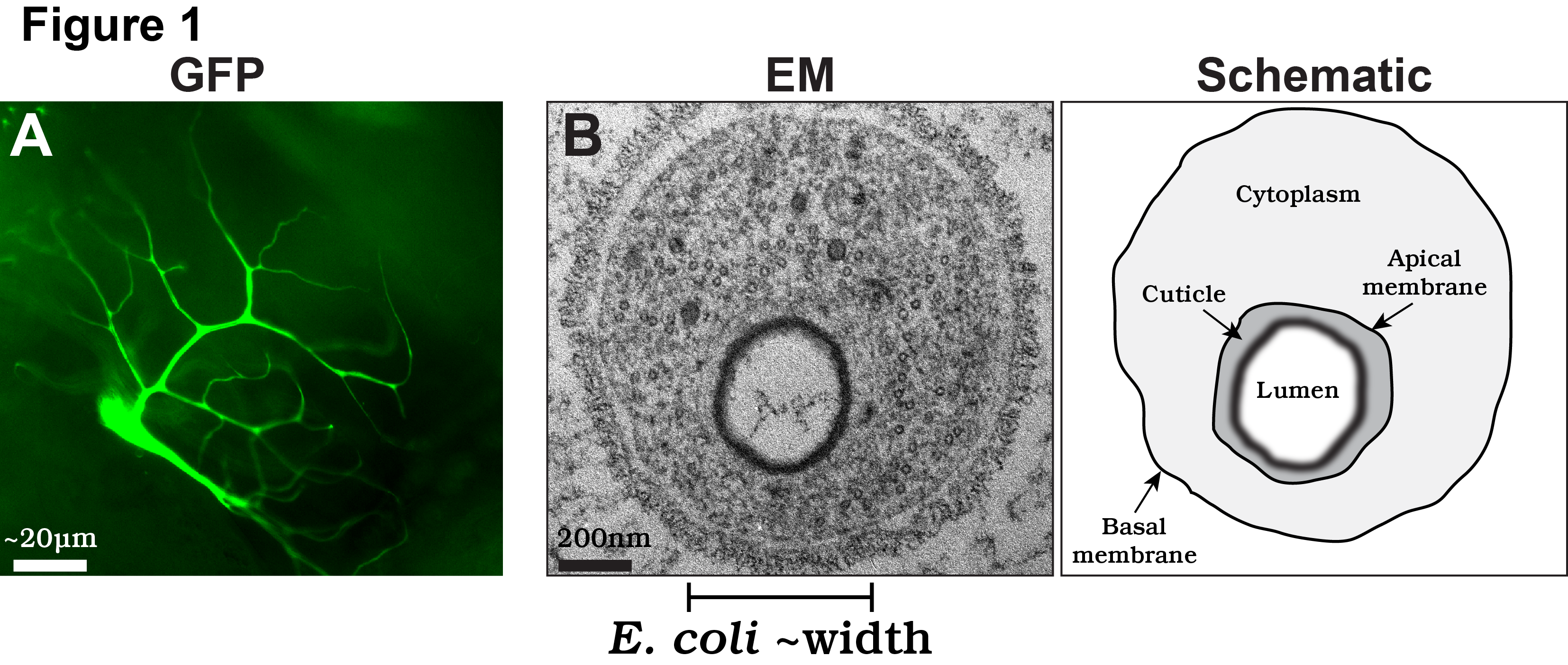
In our recent paper, we focus on the cellular and molecular mechanisms by which the subcellular lumen forms in terminal branches. The lumen of terminal cell branches is extremely thin, smaller in diameter than the width of a bacteria such as E. coli. At this scale, the formation of the lumen is akin to the mechanisms required to form subcellular organelles, such as mitochondria or lysosomes, albeit the lumen is different as it is a continuous structure extending through all the branches of the terminal cell. Despite recent amazing advances in light microscopy, the only high resolution technique available to examine the membranes that form the lumen is transmission electron microscopy (TEM; EM on left; schematized on right in Fig. 1B).
One important consideration in using EM is that the technique does not visualize biological structures directly, as the electron beam immediately vaporizes essentially all biological material. Instead, tissues must be immobilized (“fixed”) and treated with stains, typically heavy metals, that can be visualized directly in the EM. By far the most common method of fixation has been the use of chemical cross-linkers, usually aldehydes. This approach has been successfully applied to numerous samples and has produced many high quality studies. However, chemical cross-linking has some significant disadvantages. First, different biological polymers, such as membranes, proteins, or nucleic acids, differ significantly in how well they are preserved by any particular cross-linking reagent, and it is hard to find conditions in which all are simultaneously well preserved. Second, chemical cross-linking takes time, during which tissues can undergo deformation. Third, it is necessary to get the fixatives rapidly into the cells. While this is relatively easy for cells in culture and for dissected tissues, it is a significant problem for an intact organism, such as a Drosophila larvae.
To avoid the problems of chemical fixation, we turned to an alternative, very different method of fixation: high pressure freezing/freeze substitution (HPF/FS). The basic principle of HPF fixation is straightforward: cells are preserved by rapid freezing. However, as is well known, water has the unusual property of expanding upon freezing. Since most tissues are composed mainly of water, expansion from water ice crystals then causes disruption of cellular structures. However, as was proposed some 40 years ago, a procedure to avoid ice crystal damage is to rapidly freeze samples while subjecting them to high pressure. Under this regime, ice crystals cannot form. Instead the water forms an amorphic arrangement, similar to a glass. Amorphic ice has essentially the same density as liquid water, thus occupies the same volume, so damage from ice crystal expansion or tissue shrinkage does not occur. Importantly, amorphic ice maintains its structure when pressure is released, as long as the sample is kept cold. This allows the second step of the HPF/FS –freeze substitution– to proceed. During this step, water ice in the sample is replaced (substituted) with solvents that do not expand upon freezing, such as ethanol or acetone. Metal stains can be included in this substitution “cocktail” to label internal cell components. After the substitution step is completed, the samples can be returned to ambient temperature. This is followed by standard procedures of embedding in a plastic resin, sectioning, and observation by an electron microscope. Fortunately, many of the steps of HPF/FS are automated with commercial instruments available to carry out freezing and substitution (Fig. 2). Samples are frozen within a high pressure freezer, which injects liquid nitrogen at ~2500 atmospheres, both compressing and cooling the samples. Freeze substitution, which takes a number of days, takes place in a special liquid nitrogen filled chamber in which substitution cocktails are automatically exchanged, and the temperature regulated to complete the solvent exchange and the return to ambient temperatures.
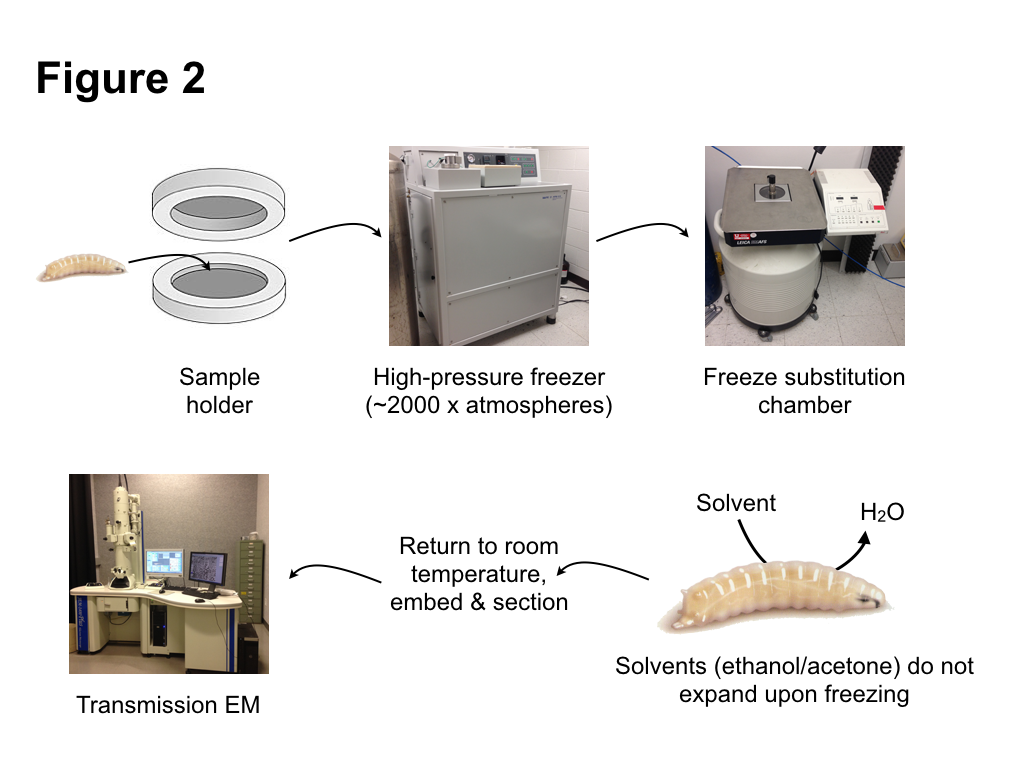
Much of the work leading to our paper involved testing different cocktails, and different incubation times and temperature change regimes during the substitution, in order to optimize both tissue preservation and structure visualization. One particularly important refinement we made during the course of our studies was the use of Durcupan instead of more commonly used epoxy resins, as it produced samples with better tissue preservation and membrane contrast. Overall, our results produced excellent preservation of internal tissues of intact Drosophila larvae. Different macromolecular structures, including proteins, nucleic acids, and the chitinous cuticle were very well preserved. Membranes in particular were particularly well preserved and had a smooth, curved appearance, indicative of very little tissue deformation.
As described in the paper, our new fixation techniques revealed previously unrecognized details of the terminal branch lumen formation. In particular, we found evidence of a hitherto undescribed intermediate of tube formation: a novel, multimembrane subcellular compartment that may contain the precursors of the cuticle lining the lumen. We also characterized the ultrastructural phenotypes of new genes required for lumen formation, providing further evidence for the multimembrane intermediate in the lumen formation process. Our future research will involve using our new fixation techniques to characterize other genes required for lumen formation and an analysis of the subcellular localization of proteins required for this process. In general, our fixation techniques should allow for analysis of many of the developmental and physiological processes that occur during Drosophila larval development.
 (2 votes)
(2 votes)2 thoughts on “High-pressure tubes”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
Love it! Elegant description of the paper. Is there a “sensor” of acidification that can be expressed in terminal cells to visualize this process and the subsequent loss of this in rabcn-3A mutants?
Thanks for your comment! We’d very much like to measure acidification. We tried dyes, but the background from other tissues was too high to be able to look at terminal branches. We’d like to try a genetically encoded sensor next. It’ll have to be targeted to the developing lumen, but it’s a good experiment to try.