BSDB Gurdon Studentship Report – Jana Hennelova
Posted by Jana Hennelova, on 2 January 2023
I have become passionate about the field of stem cell biology during my undergraduate degree in Molecular Genetics at the University of Edinburgh. Therefore, I was grateful to be awarded the Gurdon Studentship Award from The British Society for Developmental Biology to conduct a summer research project in Dr Soufi lab in the Institute for Regeneration and Repair (IRR). In this report, I will not only describe the aim of my project and how I proceeded to accomplish it, but also how valuable this experience was for my future studies and career.
In 2006, Yamanaka and Takahashi made a breakthrough in stem cell biology by showing that adult cells can be converted to become induced pluripotent stem cells (iPSCs) using the ectopic expression of four transcription factors (TFs) Oct4, Klf4, Sox2 and c-Myc (Takahashia & Yamanaka, 2006). These iPSCs are very similar to embryonic stem cells (ESCs) and have the unique ability to generate all cell types of the body. Therefore, iPSCs have enormous potential in the area of regenerative medicine. However, reprogramming adult cells into iPSCs is highly inefficient which limits the application of this technology for drug discovery and disease treatment. To improve reprogramming efficiency, further research focusing on fundamental mechanisms by which these TFs facilitate and maintain pluripotency is needed.
In my project I focused on TF Sox2, which is responsible for both somatic reprogramming and the maintenance of pluripotency in ESCs. Previous work, conducted in the Soufi lab, identified critical regions of Sox2 that are responsible for iPSC reprogramming. This was done by carrying out a systematic mutation screen of the Sox2 DNA-binding domain and mapping which regions of this domain are important for reprogramming fibroblasts to iPSCs. This led to the identification of several deletion mutants who were unable to reprogramme. My aim was to test if one of these deletions, called D36, also abolished the ability of ESCs to maintain pluripotency, which would indicate that this region of Sox2 gene is responsible for both reprogramming and pluripotency maintenance (Figure 1).
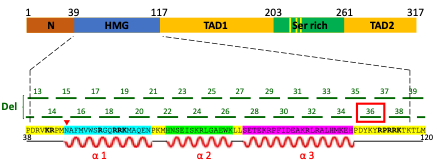
Mouse Sox2 gene is composed of functional domains including high mobility group-box (HMG) domain (i.e. DNA-binding domain), transactivation domain 1 (TAD1), serine-rich domain (Ser rich) and transactivation domain 2 (TAD2). Systematic 5 nucleotide deletions in the HMG domain led to the discovery that the D36 region (among others) is important for the function of Sox2 in somatic reprogramming. The impact of D36 deletion on pluripotency maintenance was investigated in this research project.
I used CRISPR-Cas9 technology to generate mouse ESC (mESC) lines expressing D36 Sox2. The first step in this process was to design Sox2-specific guide RNAs (gRNAs) and test their efficiency to target Sox2 gene in mESCs. This was performed by introducing a designed gRNA together with Cas9 nuclease into mESCs by electroporation, followed by alkaline phosphatase (AP) staining of mESCs. AP is one of the best markers of pluripotency (Stefkova et al., 2015) and therefore it was used determine whether these cells differentiated due to Sox2 knock-out (KO). During this phase of my project, I had to design, order, and test multiple gRNAs to find out which one was the most efficient in targeting Sox2. Ultimately, I managed to design a suitable gRNA able to mediate Sox2 KO as was demonstrated by the decreased number of AP-positive colonies after AP-staining.
Next, I performed CRISPR-Cas9 knock-in (KI) to generate mESC cell lines containing D36 deletion mutation by homology-directed repair (HDR). For that, I designed a single stranded DNA (ssDNA) template containing D36 and introduced it into mESCs together with gRNA and Cas9 complex by electroporation. Due to the low efficiency of CRISPR-Cas9 technology, single cell sorting had to be performed to identified mESCs with the mutation of interest. After that, the presence of D36 KI in the picked mESC colonies was evaluated by polymerase chain reaction (PCR) screening using D36-specific primers and confirmed by Sanger sequencing (Figure 2). Out of 38 screened colonies, one colony (colony number 27) was identified as a clear D36 homozygote, together with 6 potential heterozygotes. D36 heterozygosity arose because of low CRISPR-Cas9 efficiency, which was only able to mediate KI of only one Sox2 allele.
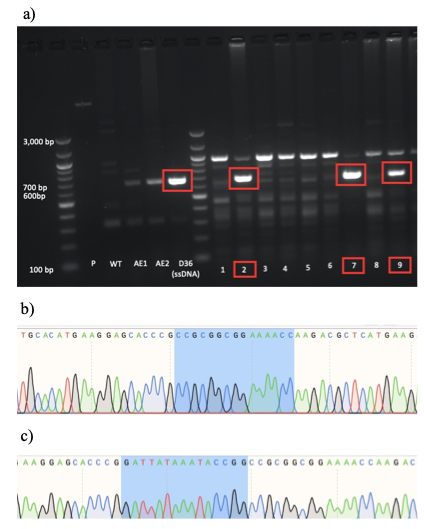
a) PCR screening followed by gel electrophoresis was used to identify D36-positive mESC colonies which produced an amplicon size of 654bp (red box). This approach led to the identification of 7 positive colonies numbered 2, 7, 9, 19, 27, 28 and 32 (note: colonies 19, 27, 28 and 32 not shown).
b) Sanger sequencing results of colony number 27, showing the clear presence of Sox2 D36 deletion (note: the highlighted region corresponds to the sequence after the D36 locus as the 15bp of the D36 deletion region are missing).
c) Sanger sequencing of WT mESCs serving as a negative control (note: the highlighted region corresponds to the locus of prospective D36 deletion, showing the presence of the D36 region).
Subsequently, the heterozygosity of the potential heterozygous colonies was validated by TopoA cloning. This approach involved the amplification of the Sox2 D36 region by PCR, its ligation into plasmid vectors and their amplification in E.coli. This allowed me to validate the zygosity of D36 mutation by sequencing individual alleles of Sox2 gene. Unfortunately, the homozygosity status of colony 27 with clear evidence of D36 mutation on both Sox2 alleles could not be validated by TopoA cloning due to the lack of time. Interestingly, there were obvious phenotypic differences between this homozygous clone and WT mESC lines (Figure 3). These differences included slow growth, smaller cell size, and greater adherence to the culturing plates. This suggests that D36 deletion most likely does have an impact on maintaining pluripotency of mESCs. Nevertheless, this important conclusion should further be validated, for example by performing embryo contribution assay to assess the ability of D36 mESCs to differentiate and generate tissues and organs.

Potential D36 homozygous mESC colony 27 on 6-well plate (a) and 12-well plate (b) had a decreased proliferative ability and smaller cell size compared to WT mESCs E14 IC passage 30 on 10cm plates (c).
During my internship, I have not only learnt different laboratory techniques, but also developed various skills and attributes that are essential for a scientific career. Most importantly, this experience helped me enhance my independent critical thinking when designing and planning experiments. Also, I was required to adjust existing protocols to suit my experiments which sometimes took multiple attempts to make it work and is therefore something I have not experienced during my undergraduate studies. Over time, I also became more confident in trusting my own judgments and making independent decisions, but also asking for help and advice when needed. In this respect, I was lucky to be a part of a stimulating scientific environment, surrounded by experienced scientists always willing to help and share their knowledge and skills.
Overall, being a part of Dr Soufi group in IRR was a very useful experience for me which also significantly influenced my future career plans. This internship cemented my aspirations to dedicate my life towards scientific research. Although sometimes difficult if things do not go as planned, it is nevertheless very rewarding and purposeful for me. Therefore, I aim to continue my research in the stem cell field or related areas that I am passionate about by doing a PhD.

References
Takahashi, K. and Yamanaka, S., (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell, 126(4), pp.663-676. Available at: https://doi.org/10.1016/j.cell.2006.07.024
Štefková, K., Procházková, J. and Pacherník, J. (2015). Alkaline phosphatase in stem cells. Stem cells international, 2015:628368. Available at: doi: 10.1155/2015/628368
 (3 votes)
(3 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.