Delineating the making of an embryo
Posted by PierreNeveu, on 15 September 2020
By Hanna L. Sladitschek and Pierre A. Neveu
Our body consists of a multitude of highly specialized tissues: the neurons in our retina seem to have little in common with the glandular cells in the intestine or our muscle fibers. Yet nothing hints at that complexity at the time of conception, when the genomes of sperm and egg fuse to define the blueprint of a new individual. Indeed, how an entire organism arises from a single cell truly is one of the most fascinating questions of biology. As the embryo develops, its building blocks (cells) gradually assume specialized functions and coordinately go through transient states that serve as intermediate steps wherefrom more complex structures will be molded as time progresses. Throughout this process, cells dynamically receive and send information to instruct their neighbors to assume specific fates. This cell-to-cell communication ensures all parts working together perfectly to create a self-organizing system that will eventually give rise to an entire body. Embryonic development thus follows a delicate choreography wherein every cell has to fulfill a precise role depending on its relative position in space and time. Unraveling the blueprint of the making of an embryo is a daunting endeavor that has fascinated developmental biologists for decades.
This study started with two physicists-turned-biologists (Pierre Neveu and Lars Hufnagel) marveling at SPIM (selective plane illumination microscopy) movies of developing Phallusia mammillata embryos, an ascidian species that is an ideal model for advanced imaging approaches due to the high optical transparency of early embryos. Our ambition was ignited to take a quantitative systems approach to decipher the molecular mechanisms underpinning the marvelous unfolding of embryonic complexity captured in those movies. We set out to employ high resolution single-cell RNA sequencing (scRNA-Seq) to decipher the complete transcriptome of every single cell in the developing embryo from zygote to gastrulation.
We quickly came to realize that the choice of an appropriate model system would be critical for the success of this highly ambitious project. Neither of us had previously worked on ascidian development but we embraced Phallusia mammillata for its many advantageous properties. Ascidian embryos are made up of a very small number of cells and their development is stereotypical. This means that the developmental history of any given cell is known but it also means that its future fate can be predicted and all individuals develop according to that same invariable blueprint. However, ascidians have a genetic makeup that resembles the ones of vertebrates (albeit less complex as genome duplication has not occurred).
We thought that possessing the complete (or near complete) collection of cells of individual embryos would enable us to ask two fundamental questions (Figure 1): could one actually determine the physical position of a given cell from its gene expression? How variable is gene expression during development?
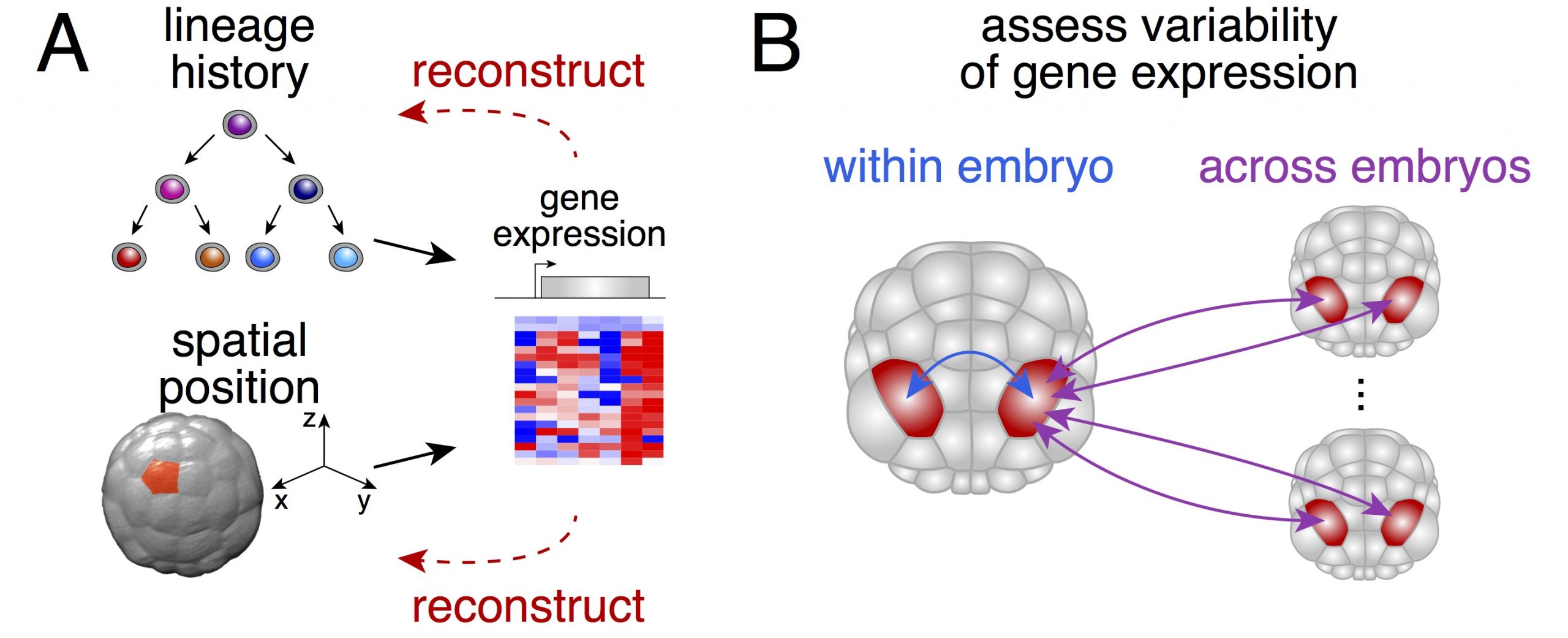
The small number of cells that make up a Phallusia embryo at gastrulation brings a crucial advantage. It allowed us to sample a large number of individuals at moderate costs in order to compare how different the gene expression is between individual embryos of the same stage.
Moreover, ascidian embryos are bilaterally symmetric so in theory there should be two copies of every cell in a given embryo (one for the left part and one for the right part of the embryo). This solves a fundamental conundrum of any single-cell approach: How to get hold of a suitable biological replicate?
Cell type identification in individual embryos
Dissociating single embryos up to gastrulation, we performed RNA-Seq at very high depth. However, a first obstacle we had to tackle was the lack of a published annotated transcriptome for Phallusia. After performing the de novo assembly of the Phallusia transcriptome, it turned out that we could capture quantitatively in single cells the expression levels of as many genes as for bulk mRNA-Seq on pools of embryos.
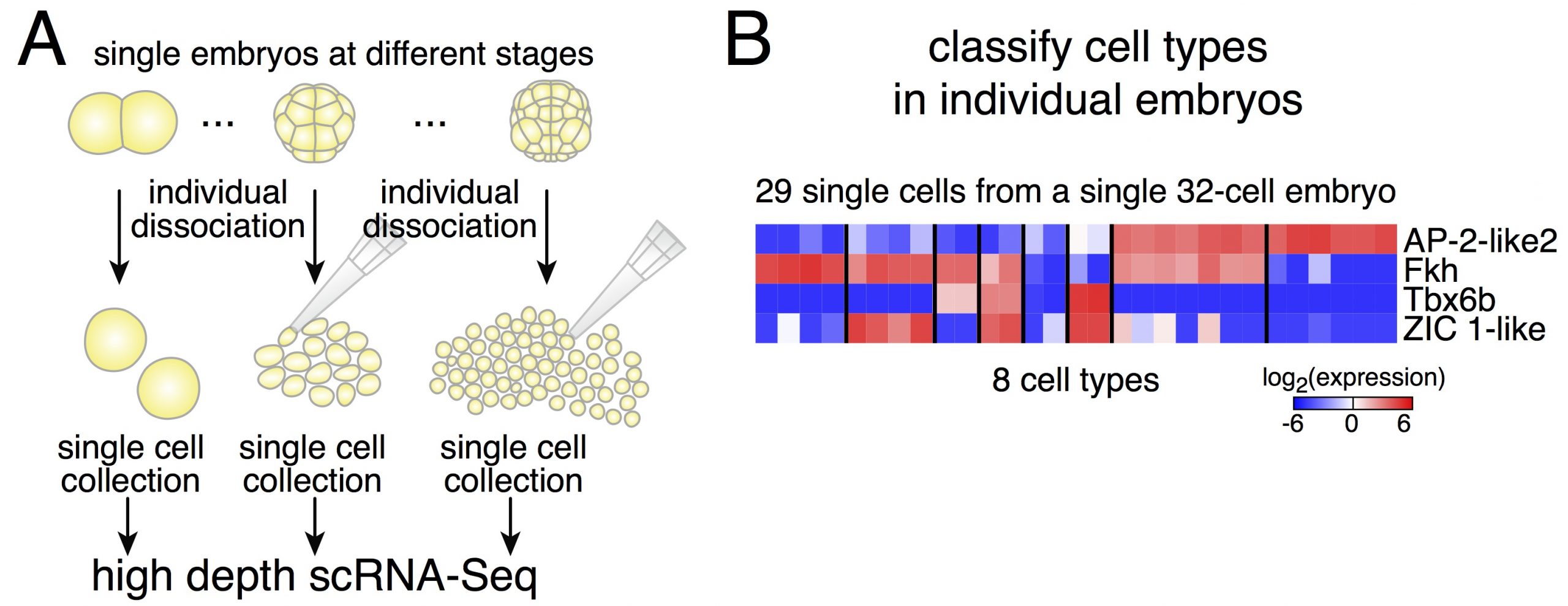
Due to potential variability between embryos, we thought it might be simpler to classify cells within a given embryo (and then compare cell types across embryos of the same stage, Figure 2). Relying on an iterative classification scheme, we identified 18 cell types in a 64-cell embryo. This diversity is striking despite the small number of cells. Another interesting point is the low dimensionality of the classification: the number of gene groups that can classify cell types is smaller than the number of cell types. It means that cell fate specification relies on the modular expression of genes (and not only on genes that would be exclusively expressed in individual cell types as what would be commonly assumed).
Reconstructing lineage trajectories from scRNA-Seq data
Equipped with the exact cell type composition of the embryo after each round of cell divisions, we then asked what is the mother-daughter relationship between cell types. When matching cell types across stages, correct accounting must factor in cell divisions, in other words there should be twice as many cells in the daughter cell types of a given mother cell type. Indeed, if one does not keep track of cell divisions, one would fail to capture offshoots of the germ cell lineage (which is itself transcriptionally silent up to the 64-cell stage and therefore stands out compared to other cell lineages).
Two findings stand out from the lineage reconstruction. First, we only found asymmetric mRNA inheritance between daughter cells in the germ cell lineage. Our dataset thus dispels the notion of mosaic development (at least relying on RNA-based factors) for all the somatic lineages. Second, specification is driven by three successive waves of upregulation of transcription factors. This stepwise cascading principle of transcription factors was valid for all somatic lineages.
Reconstructing the spatial organization of the embryo from scRNA-Seq data
Development is a dynamical process in which time and space are fundamentally linked. The gene expression of a given cell will adapt to the signals it receives from its surroundings in a history-dependent manner. Reversing the above statement, the spatial and temporal identity of every cell should be encoded in its gene expression signature. This is a crucial point as we had not kept track of the spatial origin of cells during the dissociation of the embryos. Thus, the next logical step was to see if we could reconstruct the physical organization of the embryo directly from the expression profiles of all its cells.
A perfect starting point is when the embryo activates its genome. Cells are a blank slate (except the ones that inherit maternal determinants, the future germ cells in the case of Phallusia) and will respond to the morphogenetic cues present in their environment. Expression profiles of the cells of 16-cell embryos define two axes: one separates the two embryo poles and the other one corresponds to the anteroposterior axis. Remarkably, the expression profiles of the different cells project in a way that mirrors the cells’ physical positions in the embryo. It thus appears that the future germ cells (through the inheritance of maternal determinants, among which a Wnt ligand) act as a signaling center that organizes the initial patterning of the embryo at 16-cell stage. The same map recapitulates the relative positions of the 8 cell types present in 32-cell embryos but can no longer resolve a few sister cell types at the 64-cell stage. In fact, different signaling ligands are upregulated at the 16- and 32-cell stages, changing the morphogenetic cues cells experience. The introduction of new dimensionality reduction axes at the 64-cell stage is thus necessary and theses axes map faithfully the position of the different cell types of the vegetal pole. In practice, one axis corresponds to the anteroposterior axis and the other to the medial-lateral axis.
Hence, the spatial organization of the embryo is reflected in the gene expression of individual cells and it can be reconstructed from it. Cells read out the local signaling environment and adapt their gene expression accordingly.
After assembling the Phallusia transcriptome, it became apparent that Phallusia possesses orthologs for all the transcription factors and signaling molecules expressed during embryogenesis annotated in the Ciona genome. There is a remarkable conservation of the expression territories of ortholog genes between the two species. Thus, there is a strong regulative dimension to ascidian development despite the limited number of cells constituting their embryos.
Assessing the variability and reproducibility of gene expression during development
Our dataset has both high cell coverage of individual embryos but also many embryos of a given stage. We could then study the reproducibility of gene expression within an embryo and across embryos.
How similar are the two sides of the embryo? Here again, the small number of cells in the embryo allows an unambiguous comparison. Some cell types have only two cells per embryo so there is necessarily one coming from the left side of the embryo and one from the right side. In the majority of these cell pairs originating from the same embryo, the expression of marker genes is highly reproducible with a precision comparable to the one of housekeeping genes.
While bilaterally symmetric cells in a given embryo are highly similar, there is an obvious spread in gene expression profiles within particular cell types. What could account for these differences? The most natural explanation would be that they are due to slight differences in staging. In other words, cells with lower expression levels are younger than cells with higher expression levels. An alternative explanation would be that gene expression is noisy and cells have different expression levels that do not influence normal development. However, an analysis called RNA velocity allows to extrapolate the future expression levels of a given gene by looking at the ratio between spliced and unspliced counts. RNA velocity supports the notion that cells are caught at different times along the upregulation trajectory of these genes. The coordinated upregulation of dozens of genes in a given cell type is in fact accurately modeled with a model of linear variation in time. Thus, expression differences between embryos and between cells of the same cell type can be attributed to slight differences in developmental timing. It appears that transcriptional programs guiding cell fate unfold according to a temporally well-choreographed sequence in a given cell type.
Projecting the single cell expression profiles onto a fully segmented movie of embryonic development enabled us to generate a digital embryo, i.e. a 4D representation of embryonic development accounting for every cell and every gene. This data can be explored and visualized at digitalembryo.org, where anyone can generate their own movies of the temporal and spatial gene expression dynamics oftheir genes of interest.
Charting an atlas of a developing embryo at the single-cell and whole genome level would have seemed an impossible mission a mere decade ago. The merge of developmental biology and quantitative approaches from systems biology holds great promise for the future. While traditional studies have focused on the role of individual genes in specific lineages, unraveling the complexity of the making of an entire organism will greatly benefit from the holistic perspective made available by the constant advance of single-cell techniques.
Sladitschek, H.L. et al. MorphoSeq: Full Single-Cell Transcriptome Dynamics Up to Gastrulation in a Chordate. Cell 181, 922–935.e21 (2020). doi: 10.1016/j.cell.2020.03.055
 (11 votes)
(11 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.