Mechanical cues as developmental pacers that orchestrate morphogenesis
Posted by Elias Barriga, on 22 March 2018
In these lines I share with you some details of our recently published Nature paper. I will comment how this project was started and details which are not included in the manuscript. Finally, I will briefly comment on some questions we are working on today and others we believe are worth addressing in the future.
Historical context of the project.
One of the questions that has enthralled biologists for many years is how a single cell can give rise to highly organised multicellular organisms with extremely complex forms. Although finding an answer to this question represents a big challenge, the advances made by the cell and developmental biology community over the last 50 years have been truly astounding. The advent of molecular biology in the last century, allowed embryologists to generate a strong body of evidence whereby ‘old’ ideas based upon cellular observations have found their molecular explanation. One of the most astonishing examples of this type of molecular re-description of a cellular process is the molecular characterisation of the Spemann–Mangold organiser. This tissue was initially described in 1924 as an embryonic region exerting inductive effects on cells (1). More than 70 years later, Edward De Robertis’ group generated cDNA libraries from manually dissected organisers and found genes that were specifically expressed in this territory (i.e., goosecoid (gsc)) (2). This was the first demonstration that this structure was a molecular identity defined by specific gene expression patterns. Currently, most of the embryonic tissues and about 4 organiser regions have been molecularly defined across embryos of different species (3,4). Indeed, this field has progressed to such a level that well-defined networks of signalling pathways are now widely accepted as coordinators of morphogenesis, and that their most common modus operandi is the delivery of positional information allowing interaction among neighbour cells (3,4). However, despite such vast advancements in our understanding of molecular players during morphogenesis, the role of other types of cues in this process is comparatively less understood.
Morphogenesis is a highly regulated biological process which involves constant rearrangement of tissues and deep mechanical changes. However, the role of these mechanical changes is only just re-starting to be considered when looking for a more comprehensive explanation of the morphogenetic process. I use the term ‘re-starting’ because in the early 1900’s, and prior to the molecular ’boom’, scientists studied embryonic development by using theoretical, morphological, chemical, and mechanical approaches. More recently, and ‘swimming against the tide’, several groups have focussed their research on developmental mechanics as a means of understanding those aspects of morphogenesis that molecular patterning cannot explain by itself. Work from these contemporary groups has been nicely reviewed in Lecuit, 2008 and Gilmour et al., 2016 (5,6). Considering the strong molecular frame-work and new advances in tissue mechanics, the field is starting to look at embryos as finely regulated machines where tissue rearrangements lead to mechanical changes that, in turn, feed back into the cells to modify their gene expression and behaviour, ultimately coordinating morphogenesis.
In this context, and taking advantage of powerful mechanical and molecular tools, we revealed that tissue stiffening owing to cellular rearrangements occuring in the mesoderm during Xenopus gastrulation triggers the epithelial-mesenchymal transition (EMT) and collective migration of the neural crest (NC) at neurula stages. More generally, we showed that tissue mechanics act as long-range cues coordinating the timing of two developmental processes that, until now, were considered as unconnected events (7).
Early steps of the project
The initial question addressed in this project was: what triggers the migration of the neural crest? The neural crest forms by inductive signals at the border of the neural plate (induction) where it remains as a non-migratory population until, at some point and for some reason, they undergo EMT, delaminate, and collectively migrate toward their target tissues (7,8).
The conception of this project occurred towards the end of my PhD in 2014. I was working on the transcriptional regulation of Twist, an EMT regulator in the neural crest (9). A persistent question at the time was why Twist and most of the neural crest EMT-related transcription factors are expressed at induction stages, 12 hours before the EMT and migration take place. At the same time, I was due to start a postdoc, requiring me to design and write a project. By 2012–14 there was a wave of in vitro studies showing that stiffer substrates favour migration and EMT in several cell types (10) but whether this was the case in vivo, was lacking a demonstration. That is how, with Roberto, we began to speculate that mechanics of the tissues surrounding the neural crest may change during development and that, consequently, the neural crest could feel this change and migrate. I started to read about the topic and I was impressed by the vast morphological changes that Xenopus embryos undergo in these 12 hours prior to neural crest migration, during which embryos pass from a spherical to a rather more elongated shape due to convergent extension (11,12). I even found an article showing that paraxial mesoderm stiffens towards the onset of NC migration (13). Many questions emerged at the time i.e.: Does the environment play a role in NC migration? Does NC actually reply to mechanical cues from its substrate? What are the mechanical properties of the NC microenvironment? How are these mechanical properties regulated? So, we put these questions down in a project, got the funding, and start working on this story. Not as simple as it sounds; I had to apply twice for funding, but I was finally awarded an EMBO and a Marie Curie postdoc fellowship.
Main findings
We can divide our story in two parts. First, we studied the mechanical interaction of the NC with its surroundings and the role of the temporal environment in controlling NC migration. Second, we sought to understand the mechanisms underlying mesoderm stiffening.
Before discussing the results, I would like to clarify the nomenclature we use for the different stages in which we analysed the NC. Non-migratory NC are defined as, even if it sounds obvious, cells that are not migrating and that instead resemble a more epithelial phenotype. These are normally analysed between stages 12–14 in our work. Pre-migratory NC are motile but a migratory stream is not yet observable, and we analysed them at stage 17–20. Finally, migratory NC, which are defined when migratory streams are evident, are typically analysed between stages 22–25 in our article. Stages as in Nieuwkoop and Faber (14).
I was forgetting this! When I talk about stiffness throughout the text, I am referring to the apparent elasticity of the tissue or the apparent elastic moduli contained in it, if you prefer.
Mesoderm stiffening is necessary and sufficient to trigger NC collective migration in vivo
As most of the molecules required for NC migration are already expressed at non-migratory stages, we asked whether non-migratory and migratory NC had the same migratory potential. To address this, we explanted non-migratory and migratory neural crests and evaluated their chemotactic response towards Sdf-1, a well-known NC chemoattractant (15). To our surprise, ‘non-migratory’, pre-migratory, and migratory neural crests equally migrated to SDF-1 indicating that, regardless of their stage, NC cells have the same migratory potential. Based on this in vitro observation we hypothesised that, in vivo, the temporal environment may be contributing to govern the migratory potential of the NC. Therefore, we performed heterochronic grafts, where non-migratory cells where grafted into migratory hosts and vice versa. It was nice to observe that non-migratory or migratory NC cells grafted into migrating hosts almost immediately start migrating. However non-migratory or migratory NC cells grafted into non-migratory hosts took more than 12 hours to start migrating, the same time taken by host embryos to reach migratory stages. These results strongly suggested that there is ‘something’ in the environment that changes from non-migratory to migratory stages and that this ‘something’ is very likely to be involved in the initiation of NC migration.
Personal note 1: experiments like these are highly motivating (at least for me). They do have this beautiful mixture of simplicity, creativity, and a touch of classic embryology… but with a high reward in terms of information.
As in vitro studies show that substrate stiffness triggers collective migration (10) we next characterised the mechanical environment of the NC (and yes, we did rule out other environmental factors as potential triggers of NC migration). Because the NC migrates sandwiched between the mesoderm and epidermis, we measured stiffness of these tissues in the region just in front of the NC at non-migratory, pre-migratory, and migratory stages. Our weapon of choice to measure tissue stiffness in vivo was a very novel and versatile setup of in vivo atomic force microscopy (iAFM). This method was developed by one of our co-authors, Kristian Franze (University of Cambridge) (16). After adapting this setup to our experimental requirements, we discovered that the mesoderm, used by the neural crest as a substrate, stiffens by 3 times from non-migratory to migratory stages (Figure 1). Our controls revealed that the epidermis has either similar or lower stiffness than the mesoderm. Until here, our results showed that there is a strong correlation between mesoderm stiffening and neural crest migration.
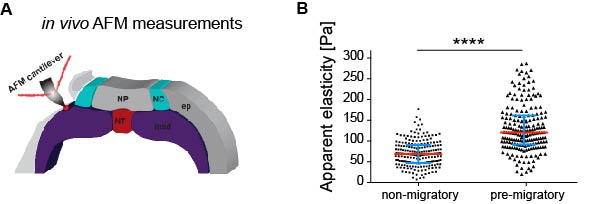
Based on this, our next question was whether this correlation plays a functional role in NC migration. We addressed this question by developing an ex vivo system consisting of 2D hydrogels containing stiffness values equivalent to those obtained in embryos at migratory (stiff) and non-migratory (soft) stages. NC plated on stiff substrates chemotax towards Sdf-1, disperse, and undergo a switch of E- to N-cadherin (EMT). However, these parameters were drastically reduced in cells plated on soft substrates. This was a nice control that showed us that cranial neural crest replies to changes in substrate mechanics.
As we learned that mesoderm stiffens and that NC behave differentially on substrates of varying stiffnesses, we tested the relevance of mesoderm stiffening for NC migration in vivo. We used a combination of ablation experiments and targeted injections of myosin contractility inhibitors to release tension and reduce stiffness in the migratory substrate of the neural crest. We controlled that our treatments effectively reduced stiffness by using iAFM. In all our conditions, NC migration was analysed by in situ hybridisation. Our results were very clear and we observed that migration of the NC was inhibited when the stiffness of the substrate was reduced by tissue ablation or targeted injection experiments (Figure 2 A–C). These results indicated that mesoderm stiffening is necessary to promote migration of the neural crest in a non-autonomous manner.
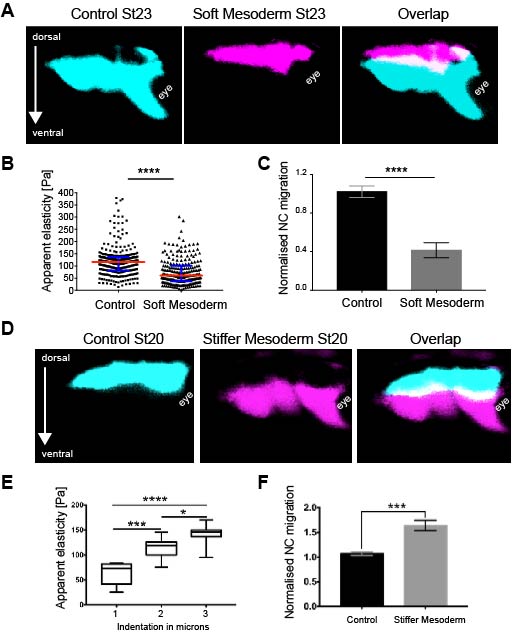
We next asked whether substrate stiffening was also sufficient to trigger migration of the NC in vivo. For this, we used a novel ‘strain stiffening’ assay to promote early stiffening of the tissue in front of the NC. This is an AFM-based method in which we can exert a controlled force for a determined amount of time in a precise location (16). As a result, we observed that applying extrinsic stress was sufficient to stiffen the mesoderm and promote premature migration of the NC (Figure 2 D–F). This was a very surprising result and it represented one of the first demonstrations of premature migration of the NC. Whilst former studies showed delamination and ectopic migration of delaminated single NC cells upon cadherin inhibition, the premature migration of the NC by following its stereotypical migratory paths had not been previously shown.
Personal note 2: designing and performing these experiments was a great experience for me. It was something not related at all to my biological background and it is one of the major sources of new knowledge from my postdoc. I have no doubt that around this time I changed the way I address biological questions.
Taking together this set of results we concluded that stiffening of the mesoderm is necessary and sufficient to promote NC migration in vivo, very likely by triggering its EMT. We believe this is not only relevant for the NC. Numerous groups of cells and single cells are challenged by the mechanics of their surroundings when migrating in vivo. There are several types of mechanical challenges that cells need to sort in order to reach their target tissues. Hence it would be of great value to start exploring the mechanical interaction of other cellular systems with their surrounding tissues in diverse biological contexts.
PCP-mediated convergent extension is the driving force of mesodermal stiffening in vivo
Our next goal was to unveil the driving force of mesodermal stiffening. The first idea we explored was an accumulation of extra cellular matrix (ECM). In many systems, accumulation of ECM is one of the main components of tissue stiffening (17). It was disappointing to discover that in our system this could not be the case. We found out that i) there is no difference in ECM deposition among non-migraotry and migratory stages; ii) removing ECM under the NC did not affect mesoderm stiffness; and iii) collagen is not expressed at these stages in Xenopus.
Thus, we next turned our attention to actomyosin contractility, as it has been previously shown that this activity could play a role in paraxial mesoderm stiffening (13). We analysed expression of phosphorylated myosin and f-actin in the head lateral mesoderm (mesoderm used by the NC as a substrate). We observed no differences in expression levels or sub-cellular localisation when comparing non-migratory vs migratory stages. We even inhibited contractility once the stiffness was already increased in the head mesoderm. As measured by iAFM, only a very small and non-significant difference was observed when compared to control embryos, indicating that although it may play a role, contractility was not the main component of head mesoderm stiffness.
Personal note 3: These were very frustrating results at the time, I was running out of options and the explanation about what was the driving force of mesodermal stiffening seemed more complicated than what we thought. But if you ask me today I think that this complication is from where one of the most interesting parts of our discovery was originated; mechanical coordination among temporally unrelated embryonic tissues.
We did not give up and kept looking for options. At the time, Kristian’s lab was just publishing their work about differential cell density as the source of a stiffness gradient that guided the growth of the optic tract (16). We went back and looked into the anatomy of the embryo and realised that from non-migratory to migratory stages, embryos elongate and cells accumulate dorsally. We did a very simple sectioning experiment on wild-type embryos with colour-labelled mesoderm and NC. We quantified cell density in the mesoderm under the NC at non-migratory, pre-migratory, and migratory stages and observed a strong accumulation of cells and increased cell density that nicely correlated with mesoderm stiffening and onset of NC migration (Figure 3 A–C).
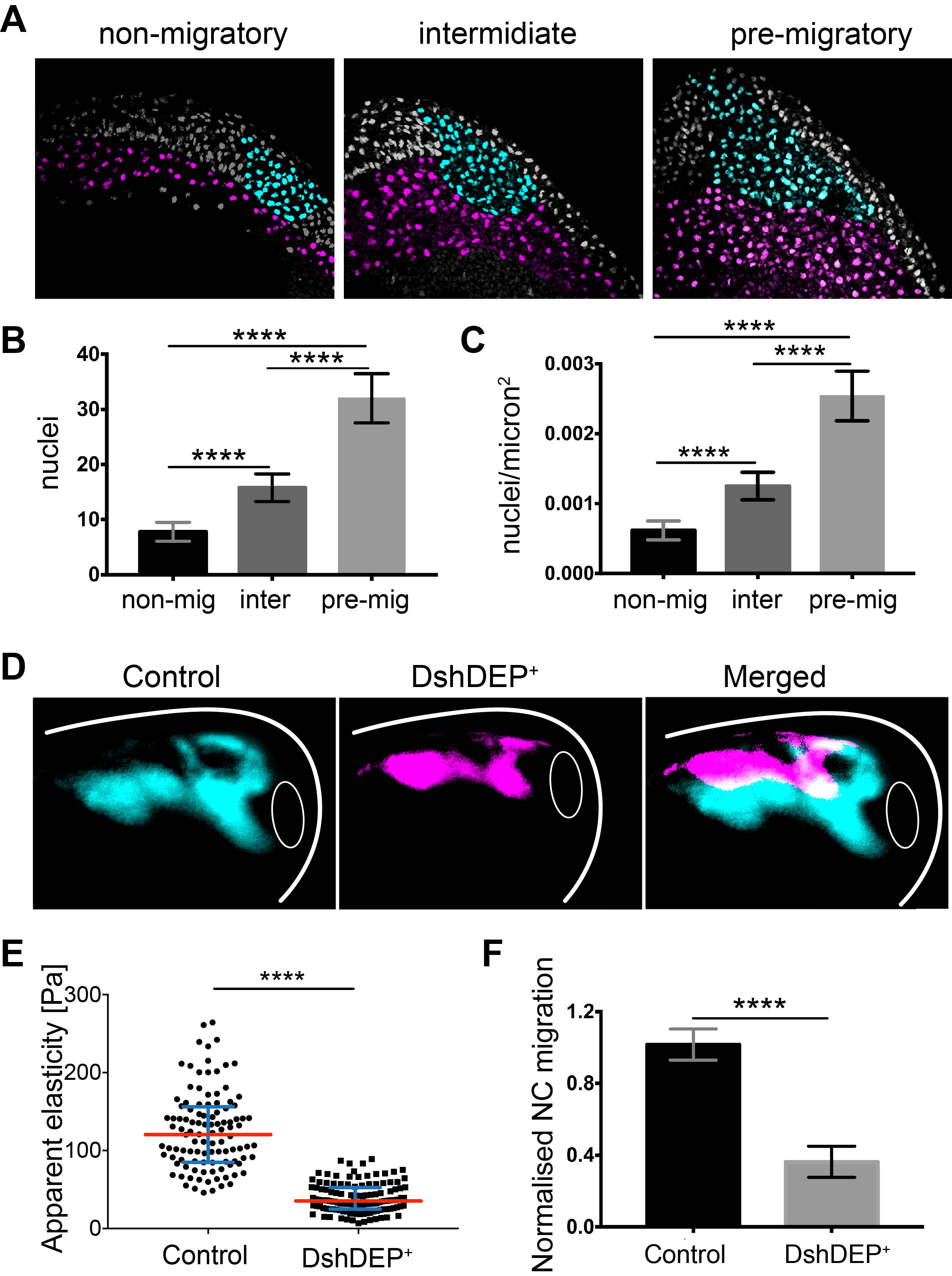
To functionally test this correlation, we quantified cell density upon application of distinct treatments into the mesoderm without inducing a direct effect on NC migration. We found that treatments resulting in blocked NC migration correlated with low mesodermal cell density and low stiffness, whereas treatments promoting NC migration perfectly corresponded with high mesodermal cell densities and high stiffness values. This strongly suggested that one of the main components of mesodermal stiffness is the accumulation of mesodermal cells that lead to increased cell density.
This matched seamlessly with what we observed before and at this point we re-interpreted our myosin knockdown results, where early inhibition of myosin contractility in the mesoderm led to decreased stiffness and inhibited NC migration. It is well-known from John Wallingford’s lab work that polarised activity of myosin is required for mesodermal cell migration (12). Consequently, it makes sense to think that what our treatments were doing was inhibiting mesoderm cell migration which in consequence decreased cell density and stiffness under the NC.
As planar cell polarity (PCP) is a master regulator of convergent extension (CE) (11,12) we inhibited PCP in the mesoderm. As expected, mesoderm stiffness was decreased and NC migration inhibited (Figure 3 D–F). As depleting PCP has many biological consequences, this treatment represented an excellent scenario to test whether introducing extrinsic stress to stiffen the mesoderm of soft embryos was enough to promote NC migration. So, as a final experiment we inhibited PCP, applied extrinsic stress with iAFM and observed that NC migration was mechanically rescued (Figure 4). These results led us to conclude that the driving force behind the increased cell density that promotes mesoderm stiffening and NC migration are PCP-driven cellular rearrangements during CE, with a minor contribution of myosin contractility.
Personal note 4: Something which I believe is really worth mentioning is the role of myosin and ECM in mesoderm stiffening. There is a large body of evidence from excellent groups that demonstrate the requirement of Fibronectin deposition and myosin activity to ensure correct convergent extension movements. Hence myosin and Fibronectin do contribute to build-up mesodermal stiffening by allowing mesoderm migration and accumulation of cells under the NC, but they represent a minor contribution to stiffness once cells have already accumulated.
These results were very exciting as they demonstrated for the very first time that two embryonic processes that have been always studied as separate events, CE during gastrulation and NC migration, are connected via tissue mechanics. Our data is an example of how applying more integrative approaches to study morphogenesis can lead us to actually understand how the process organises itself– such an approach has the potential to deeply impact our advances in the study of ‘self-organising’ biological systems.
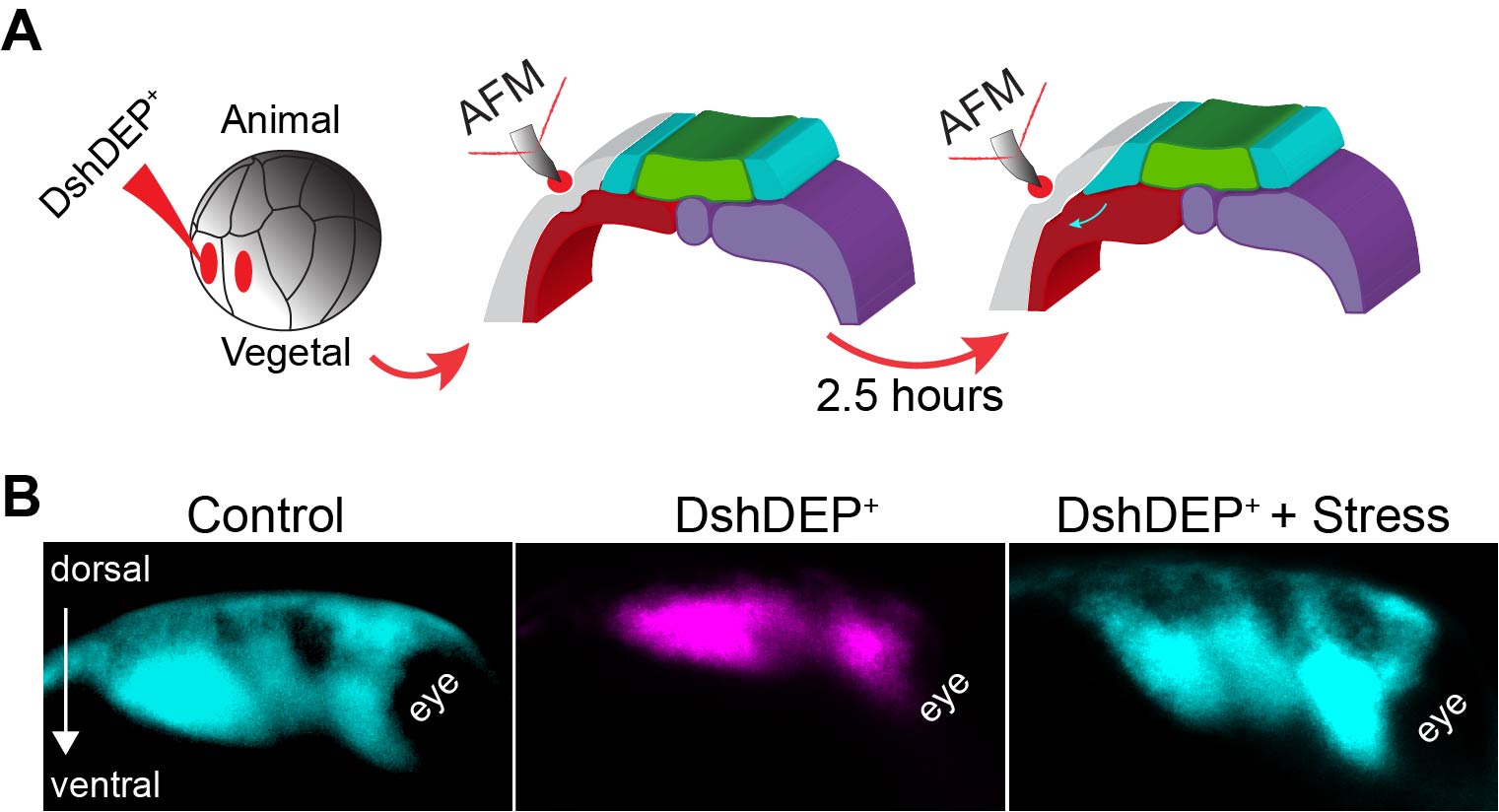
Figure 4. Ectopic compression rescues NC migration in soft embryos. A Diagram describing the extrinsic stress experiments used to stiffen the mesoderm after PCP inhibition. B Representative images showing lateral views of embryos hybridised with a probe against a NC marker. Treatments as indicated.
Open questions
Although we demonstrate in our article that integrin, vinculin and talin, well-known mechanosensors (18), are required for NC migration we are now actively studying the mechanism by which NC cells sense and translate mechanical stimuli in a cellular and eventually molecular response. Another question that we are addressing now is related to a potential role for durotaxis in vivo. Durotaxis is the process by which cells migrate from compliant to stiffer substrates. In this front, we are studying how stiffness gradients may emerge as a result of mechanical heterogeneities of embryonic tissues and whether these gradients can direct cell migration in vivo. In the future, it may be interesting to address whether substrate mechanics are just permissive cues allowing cells to exert traction and migrate or instead are a real source of signalling that ‘permit’ migration but also modify the genetic program of the NC. Similarly, a question that may be linked to environmental mechanics and that is relevant for development, regeneration, and cancer is how cells know when to stop migrating and differentiate once they reach their target tissues.
Acknowledgements
I would like to thank the co-authors of this article Roberto Mayor, Guillaume Charras, and Kristian Franze for their input and the funding sources EMBO (LTF-971) and Marie Skłodowska Curie (IF-2014_ST VivoMechCollMigra). Dr Ailin Buzzi (KCL) and Emily Atalar (UCL) for their comments on this post.
References
1. Spemann H, & Mangold H. Induction of embryonic primordia by implantation of organizers from a different species (1924). Roux’s Arch. Entw. Mech. 100, 599–638.
2. Cho, K. W., et al., (1991). Molecular nature of Spemann’s organizer: The role of the Xenopus homeobox gene goosecoid. Cell, 67, 1111–1120.
3. Anderson, C. & Stern, C. D. (2016). Organizers in development. Curr. Top. Dev. Biol. 117, 435–454.
4. De Robertis EM (2006). Spemann’s organizer and self-regulation in amphibian embryos. Nat Rev Mol Cell Biol. 7, 296–302.
5. Thomas Lecuit. (2008). “Developmental mechanics”: cellular patterns controlled by adhesion, cortical tension and cell division. HFSP Journal 2, 72-78.
6. Gilmour, D., Rembold, M. & Leptin, M. From morphogen to morphogenesis and back. Nature 541, 311–320 (2017).
7. Barriga E.H., et al., (2018). ‘Tissue stiffening coordinates morphogenesis by triggering collective cell migration in vivo’. Nature 554, 523–527.
8. Betancur, P., Bronner-Fraser, M. & Sauka-Spengler, T (2010). Assembling neural crest regulatory circuits into a gene regulatory network. Annu. Rev. Cell Dev. Biol. 26, 581–603.
9. Barriga, E. H., et al., (2013). The hypoxia factor Hif-1α controls neural crest chemotaxis and epithelial to mesenchymal transition. J. Cell Biol. 201, 759–776.
10. Roca-Cusachs, P., Sunyer, R. & Trepat, X. Mechanical guidance of cell migration: lessons from chemotaxis. Curr. Opin. Cell Biol. 25, 543–549 (2013).
11. Wallingford J.B. & Harland R.M. (2001). Xenopus Dishevelled signaling regulates both neural and mesodermal convergent extension: parallel forces elongating the body axis. Development 128, 2581–92.
12. Shindo A. & Wallingford J.B. PCP and septins compartmentalize cortical actomyosin to direct collective cell movement. Science 343, 649–652 (2014).
13. Zhou J., Kim H. Y. & Davidson L. A. (2009). Actomyosin stiffens the vertebrate embryo during crucial stages of elongation and neural tube closure. Development 136, 677–688.
14. Nieuwkoop, P. D. & Faber, J. Normal Table of Xenopus laevis (Daudin): a Systematical and Chronological Survey of the Development from the Fertilized Egg till the End of Metamorphosis 2nd edn (North-Holland, 1967).
15. Theveneau, E. et al (2010). Collective chemotaxis requires contact-dependent cell polarity. Dev. Cell 19, 39–53.
16. Koser, D. E. et al (2016). Mechanosensing is critical for axon growth in the developing brain. Nat. Neurosci. 19, 1592–1598.
17. Moeendarbary, E. et al. (2017). The soft mechanical signature of glial scars in the central nervous system. Nat. Commun. 8, 14787.
18. Charras, G. & Sahai, E. (2014). Physical influences of the extracellular environment on cell migration. Nat. Rev. Mol. Cell Biol. 15, 813–824.
 (16 votes)
(16 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.