Mitotic Bookmarking by Esrrb: an unexplored mechanism for implementing cell fate choices?
Posted by Nicola_Festuccia, on 18 November 2016
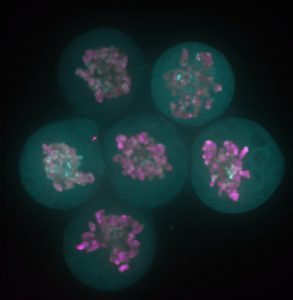
The story behind our recent paper (Festuccia et al., Nature Cell Biology 2016) started with a serendipitous observation made in a small room of the ISCR (Institute for Stem Cell Research), which at the time was still located in the King’s Buildings campus, south of town in my beloved Edinburgh. I was imaging a newly generated reporter ES cell line expressing a fusion between my favourite pluripotency transcription factor, Esrrb (Estrogen Related Receptor Beta), and a bright red fluorescent protein, TdTomato, when I noticed something I didn’t expect: I was able to identify dividing cells just by looking at the fluorescence signal from the Esrrb-TdTomato reporter. Esrrb was painting the mitotic chromosomes. I was surprised, and at the same time I realised I knew very little about the behaviour of transcription factors during cell division. Were all transcription factors supposed to remain bound to chromatin after condensation? And if so, with what consequences? Was this phenomenon reported in ES cells? What about other pluripotency factors?
Since then – I was finishing my PhD in the group of Ian Chambers – 4 years passed, and I learned that I was observing a phenomenon already described as “mitotic bookmarking”. Pioneering work over the last two decades in the groups of David Levens, Kenneth Zaret, Gary Stain, Marco Pontoglio and Gerd Blobel, among others, helped us understand that some proteins, including sequence specific transcription factors, remain bound to the chromatin during mitosis, marking promoters and key regulatory regions to facilitate the timely reactivation of gene expression after cell division. At the time of the observation, a friend and Post-Doc in Ian’s lab, Pablo Navarro, drove my attention to this concept. Pablo had already attempted to document bookmarking in ES cells. Unfortunately, regular fixatives such as formaldehyde delocalise transcription factors from mitotic chromatin (Pallier et al., Molecular Biology of the Cell 2003). The stimulating environment created by our usual scientific discussions made it possible for the implications of my initial observation not to remain unexplored. Thanks to the generosity of Ian, I was able to make of that observation the basis for my post-doc project, and I moved to the Pasteur Institute in Paris to join the newly established lab of Pablo. In these last 3 years, we were able to show that indeed during mitosis Esrrb remains bound to crucial enhancers regulating ES cell identity and instructs gene expression in daughter cells. We also understood that mitotic binding is very dynamic, and chiefly driven by the interaction of Esrrb with its consensus binding sites on DNA.
Nonetheless, many of the question I had at the time of the first observation of Esrrb mitotic retention remain open: the challenge that cell division, and another potentially disruptive phase of the cell cycle, DNA replication, pose to the control exerted by pluripotency factors over ES cell identity is a largely overlooked problem in our field. More generally, our understanding of the properties and structural organisation of mitotic, or newly replicated, chromatin is superficial. Can the observation made for Esrrb be extended to other pluripotency regulators? In the case mitotic binding will prove to be a general phenomenon, the current assumption of a restricted accessibility of mitotic chromatin will have to be challenged. Indeed, recent observations indicate that accessible genomic regions are largely invariant between interphase and mitosis (Hsiung et al., Genome Research, 2015). Coupled to the notion that a major portion of mitotic chromosomes is not constituted by chromatin (Booth et al., Molecular Cell 2016), this indicates that part of the DNA might reside outside domains formed of compacted nucleosomes in mitosis, possibly maintaining full or increased accessibility to transcriptional regulators, chromatin modifiers and remodelers. Or, vice-versa, we could imagine that precisely the binding of transcription factors keeps portions of the genome decompacted and localised in accessible regions of the chromatids.
If ES cells use specific mechanisms to ensure the continuity of transcription factor control over gene expression during cell division, and given that the activity of pluripotency regulators, like Esrrb, is tightly linked to the self-renewal ability of ES cells, could it be that the constraints posed by mitosis make it a favourable moment for ES cells to trigger differentiation programmes? Mitotic binding by Esrrb is restricted to only a fraction of the interphase targets. Declining levels of Esrrb and other factors might therefore trigger the further constriction of the set of accessible enhancers during cell division, irreversibly destabilising the pluripotency network in daughter cells. In fact, as part of my PhD research, I observed that Esrrb downregulation marks ES cell commitment to differentiation, and identified a set of enhancers that is inactivated during the early stages of this process (these results form the basis of a manuscript currently under revision). Many of these are indeed bookmarked enhancers.
I might just find myself in the lucky position of being able to link two parts of my previous work and contribute to our better understanding of how cell fate decisions are implemented by pluripotent cells early during development, and possibly later on in multipotent progenitors and adult stem cells.
 (11 votes)
(11 votes)One thought on “Mitotic Bookmarking by Esrrb: an unexplored mechanism for implementing cell fate choices?”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
Great story indeed, I hope to hear more about mitotic bookmarking soon!