3D retinas made from patient stem cells shed light on mechanisms of inherited blindness
Posted by Amelia85, on 15 July 2016
Amelia Lane, David A. Parfitt, Conor M. Ramsden, Peter J. Coffey, Michael E. Cheetham
Parfitt et al. Cell Stem Cell 1–13 (2016)
Making eyes
Methods to differentiate human stem cells into retinal cell types have been under development for almost a decade. Stem cell derived retinal cells provide a rich resource to study unique and otherwise inaccessible cells. Reprogramming somatic cells into iPSC allows us to study retinal cells carrying pathogenic mutations that cause retinal degeneration. This enables the interrogation of disease mechanisms and allows us to screen mutation-specific therapies such as antisense oligonucleotides on patient cells. The substantial technical challenges of consistently and uniformly generating terminally differentiated photoreceptors are gradually being overcome allowing us to make use of this new and powerful experimental paradigm.
The retina is derived from the anterior neural plate. During vertebrate eye development a common pool of progenitor cells invaginate to form a double walled optic cup. The cup is patterned by interactions with the adjacent surface ectoderm to form neural retina (NR) and an overlying monolayer of pigmented cells known as retinal pigment epithelium (RPE) (Fuhrmann et al., 2000). Mature photoreceptors in the human retina have a unique structure suited to their purpose as sensors that transduce light into an electrochemical signal. Rod and cone cells have distinct compartments. Their cell bodies are arranged in an outer nuclear layer (ONL), consisting of tightly packed columns approximately 10 nuclei in thickness. At the apical edge they have tight junctions forming an outer limiting membrane (OLM). Above the OLM are inner-segments (IS), which house much of the ER and Golgi and are densely packed with mitochondria. Protruding from the IS of each photoreceptor cell is a specialised cilium, the outer segment (OS), a 25um stack of membranous discs packed with photosensitive opsin proteins and the phototransduction machinery. The OS is joined to the IS by a transition zone referred to as the connecting cilium (CC).
The first differentiated retinal cell type to be derived from human embryonic stem cells (hESC) were RPE, the cells which overlay the photoreceptors in the retina. Pigmented RPE were first detected following spontaneous differentiation of confluent hESC cells (Klimanskaya et al 2004). It has since been demonstrated by several labs around the world that induced pluripotent stem cells (iPSC) and hESC will spontaneously differentiate into RPE within a month. Making photoreceptors requires longer periods of time similar to the process in utero. In the first published attempts at photoreceptor differentiation, hESC cells were treated with Wnt and nodal inhibitors followed by long term treatment (more than 130 days) with retinoic acid and taurine (Osakada et al., 2008, Lamba et al., 2006). These protocols generated immature photoreceptor progenitor cells and/or opsin-expressing cells with no specific morphological resemblance to photoreceptors in a two-dimensional format.
Pioneering work by the Sasai lab at the RIKEN Centre for Developmental Biology in Japan, using a three dimensional ‘organoid’ approach for cell differentiation, greatly improved the quality of the photoreceptor cells derived. First with mouse, then with hESCs, Sasai’s lab was able to create photoreceptors from embryoid bodies (EBs) that formed three-dimensional (3D) ‘optic cups’ with RPE and several neural retina layers (Eiraku et al., 2011, Nakano et al., 2012). This was rapidly followed by similar demonstrations in human iPSC (Zhong et al., 2014, Reichman et al., 2014). These opsin-expressing photoreceptor cells developed mitochondria-rich inner segments with cilia easily observed, raising the tantalising possibility that they may be capable of generating OS in vitro.
Disease modelling
Our lab’s research interest in inherited retinal dystrophy disease mechanisms prompted us to attempt to make 3D retinas from cells donated by a patient with a type of retinal degeneration known as Leber congenital amaurosis (LCA). LCA is a recessively inherited retinal dystrophy resulting in severe visual loss in early childhood. This patient is homozygous for a deep intronic mutation in the gene encoding CEP290 (c.2991+1665A>G), which is the most common LCA associated allele. CEP290 is found at the transition zone of all ciliated cells and in the CC of photoreceptors. Its position indicates a role in formation of the Y-shaped linker structures that anchor the ciliary axoneme to the plasma membrane. Mutations in CEP290 can cause syndromic ciliopathy involving several organs or LCA alone. The common c.2991+1665A>G mutation is only associated with LCA and leads to mis-splicing of the CEP290 transcript and inclusion of the stop-codon containing cryptic exon. Some correctly spliced transcript remains and it is thought that the residual protein level is sufficient to prevent a syndromic ciliopathy in affected individuals. Fibroblasts and lymphoblast cultures derived from these patients display a cilia defect; with reduced numbers of shortened cilia (Collin et al., 2012, Gerrard et al., 2012, Garanto et al., 2016). In patients however, it is not clear why retinal cells appear to be affected more than other tissues.
Our first attempts at the 3D organoid method used embryonic stem cells (ESC) and control iPSC derived from neonatal foreskin fibroblasts. The ESC EBs generated very promising bi-laminated optic cups with retinal ganglion and neuroblastic cell layers as well as RPE. The neuroblastic layer contained photoreceptor progenitors, but we did not continue the differentiation for long enough to generate mature photoreceptors with IS.
We then began large scale and long-term differentiations using both control and CEP290 patient derived iPSC lines. We tried both Zhong and Nakano protocols from several patient and control lines and carried out immunocytochemistry (ICC) and gene expression analysis to follow the time course of photoreceptor differentiation.
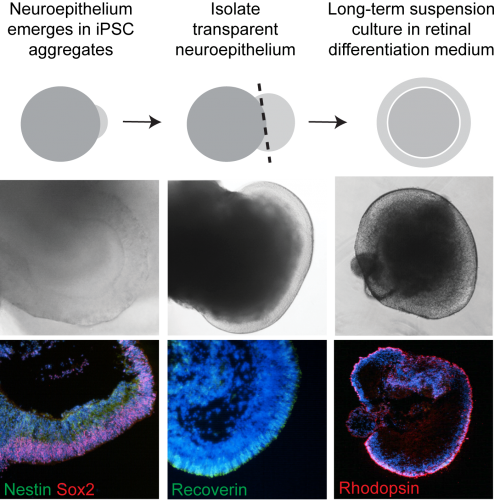
CEP290 patient fibroblasts reprogrammed efficiently into iPSC. Similar to the fibroblast cells, there was a significant reduction in ciliation in iPSC; however, this did not appear to affect their pluripotency. CEP290 iPSC efficiently differentiated into early derivatives of all three germ layers. We found that different clonal lines from both control and patient iPSC formed EBs with varying efficiency (50-100%). Of these, a proportion could be seen to generate transparent, radially aligned neuroepithelium (NE) – which again varied between lines (5-30%). We were able to distinguish these NE-producing EBs morphologically with a light microscope and dissect out the neuroepithelial buds (Figure 1). Unfortunately, a proportion of these did not survive the dissection procedure or spontaneously collapsed and became necrotic. However, the precious surviving organoids were cultured for up to 21 weeks.
We processed EBs early on in the differentiation process and saw that the basal bodies and associated cilia had aligned at the apical surface of the EBs with neuroepithelial domains, thereby fulfilling their roles as sensors that detect and relay signals from the outside environment, and facilitating polarised cell division. Excitingly we detected a phenotype in our CEP290 EBs – significantly fewer of the pericentrin positive basal bodies at the apical surface possessed ARL13B positive cilia. This did not appear to reduce the efficiency with which CEP290 EBs generated organised neuroepithelial domains; an interesting observation given the role of primary cilia in embryonic forebrain development (Willaredt et al., 2013).
We extracted RNA from individual EBs and analysed gene expression as well as cryosectioning them for ICC analysis. As the EBs developed it became clear that the differentiating cells were following the highly conserved sequence of eye development; similar to what has been observed in other studies of iPSC-retina differentiation (Meyer et al., 2009). Various photoreceptor cell markers such as recoverin and arrestin were being switched on over time (Figure 1). Comparing the time course over which markers were expressed it was clear that the patient cells were behaving similarly to controls and our model did not display any ‘developmental’ defects in vitro.
We used electron microscopy to analyse the ultrastructure of these developing photoreceptors; at week 13 tight junctions were visible all across the apical layer forming an OLM and above this mitochondria rich buds had appeared, with the morphology of inner segments.
At the latest time point (21 weeks), ICC revealed the presence of rhodopsin positive rods (Figure 2), as well as red/green and blue cone cells . These were arranged within a compacted apical layer, reminiscent of the ONL and approximately 5 nuclei thick. By this time, the interior of the cups was largely empty or necrotic. RPE cells could also be seen in several of the cups but it was generally adjacent to the photoreceptors cells and very rarely overlaying them as in the 2 walled optic cup seen in human development and reported by Sasai’s lab (Nakano et al., 2012).
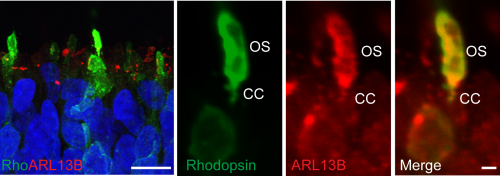
Looking at this latest time point by electron microscopy we were astonished to see the presence of connecting cilia that were elaborating into partially stacked discs similar to rudimentary OS. The discs were aligned at the base but disorganised and broken at the tip. In vivo the outer segments are enveloped by the microvilli of the RPE and encased in an extracellular matrix with a unique composition known at the interphotoreceptor matrix (IPM) (Ishikawa et al., 2015). This proteoglycan-rich substance is secreted by both the RPE and the photoreceptors in vivo and, in contrast to the majority of ECM in other tissues, is lacking in a collagenous meshwork with neither laminin nor fibronectin as major components. We observed a lot of broken cilia and opsin positive debris at the apical edge and hypothesise that if we could place the developing photoreceptors in an environment that would stabilise the nascent OS, akin to the IPM, we might observe more organised and abundant OS structures. This could be a powerful tool for studying the numerous mutations in retinal dystrophy genes that lead to OS trafficking disorders and retinal degeneration.
There was a reduction in cilia number in CEP290 patient optic cups using ARL13B to mark the connecting cilia of opsin positive photoreceptors. In addition the connecting cilia were now significantly shorter than those in controls. However the most interesting insight came from looking at the relative levels of mis-splicing in the various cell types derived. While the ratio of correctly spliced: mis-spliced transcript in fibroblast, iPSC and iPSC-RPE was approximately 50:50, in photoreceptors the quantity of correctly spliced transcript was severely reduced down to only 10-20%. Cell type specific levels of mis-splicing could explain the specificity of retinal involvement characteristic of this particular mutation, with less full-length CEP290 protein in the retina than in other tissues. The reasons for this different mRNA processing are currently unclear, but it was recently shown that the human retina has unexpectedly high levels of splicing diversity (Farkas et al., 2013) and mutations in splicing factors can cause dominant retinal dystrophy. This finding raises the possibility that the retina could be especially sensitive to intronic variants that affect splicing.
Drug testing
The ability to make human retinal cells in vitro opens up lots of possibilities to study human development, disease mechanisms and test novel therapies. Antisense oligonucleotides (AON) that bind to and sterically block the aberrant splice donor site in the mutated CEP290 pre-mRNA have been developed and tested in patient fibroblasts (Collin et al., 2012 Gerrard et al., 2012). We were able to show that 10mm of an antisense morpholino (MO) specific to the LCA CEP290 mutation (CEP290-MO) transfected in EndoPorter solution (from Genetools) reduced the amount of cryptic exon containing-mRNA produced and increased ciliation in patient fibroblasts. We also showed that the treatment lasts up to 6 days.
Testing this potent MO on the CEP290 optic cups was the next logical step. In order to assess the capacity of the MO to enter the optic cup we used a fluorescein-tagged MO and imaged all layers of the cup 48 hours later using live confocal microscopy. The tagged MO was able to penetrate all layers of the optic cup and access the nuclei. Therefore, we treated 90-day-old patient optic cups with CEP290-MO every 3-4 days for 4 weeks and then assessed cilia length, cryptic exon expression, CEP290 levels and the amount of various CEP290 interaction partners at the basal body. CEP290-MO treatment significantly increased CEP290 protein levels, reduced cryptic exon expression and all but restored cilia numbers and length to wild type levels. Critically it also restored the localisation of the important CEP290 interacting partner, RPGR, to the connecting cilia. RPGR is located in the same ciliary compartment as CEP290 and mutations in RPGR are a major cause of retinitis pigmentosa (Breuer et al., 2002). We used high magnification confocal stacks to compare RPGR and CEP290 fluorescence at the ciliary base. We observed a significant depletion of RPGR and CEP290 in patient optic cups and a significant restoration following CEP290-MO treatment, an important functional read-out for the treatment efficacy.
In the absence of an animal model that accurately recapitulates the mis-splicing mutation, the patient iPSC-photoreceptor technique has yielded unique and important mechanistic insights and provided an explanation as to why this most common CEP290 mutation leads to a retina-only phenotype in human cells. In addition we have demonstrated the efficacy of AONs in rescuing correctly spliced CEP290 levels. Hopefully these studies will encourage the use of iPSC-derived organoids to study disease mechanisms and facilitate the development and clinical application of new therapies.
Breuer DK, Yashar BM, Filippova E, et al. A Comprehensive Mutation Analysis of RP2 and RPGR in a North American Cohort of Families with X-Linked Retinitis Pigmentosa. American Journal of Human Genetics. 70(6),1545-1554 (2002),
Collin, R. W. et al. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther. Nucleic Acids 1, e14 (2012).
Eiraku, M. et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 472, 51–56 (2011).
Farkas, M. H. et al. Transcriptome analyses of the human retina identify unprecedented transcript diversity and 3.5 Mb of novel transcribed sequence via significant alternative splicing and novel genes. BMC Genomics 14, 486 (2013).
Fuhrmann, S., Levine, E. M. & Reh, T. a. Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development 127, 4599–609 (2000).
Garanto, A. et al. In vitro and in vivo rescue of aberrant splicing in CEP290 -associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. (2016).
Gerard, X. et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol. Ther. Nucleic Acids 1, e29 (2012).
Ishikawa, M., Sawada, Y. & Yoshitomi, T. Structure and function of the interphotoreceptor matrix surrounding retinal photoreceptor cells. Exp. Eye Res. 133, 3–18 (2015).
Klimanskaya, I. et al. Derivation and Comparative Assessment of Retinal Stem Cells Using Transcriptomics. Cloning Stem Cells 6, 217-245 (2004).
Lamba, D. a, Karl, M. O., Ware, C. B. & Reh, T. a. Efficient generation of retinal progenitor cells from human embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 103, 12769–74 (2006).
Meyer, J. S. et al. Modeling early retinal development with human embryonic and induced pluripotent stem cells. Proc. Natl. Acad. Sci. U. S. A. 106, 16698–703 (2009).
Nakano, T. et al. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 10, 771–85 (2012).
Osakada, F. et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat. Biotechnol. 26, 215–24 (2008).
Willaredt, M. A., Tasouri, E. & Tucker, K. L. Primary Cilia and Brain Development. Mech. Dev. 130, 373–380 (2013).
 (3 votes)
(3 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.