Adventures in Studying Brain Sex Differences
Posted by BNugent, on 8 May 2015
by Peg McCarthy and Bridget Nugent
The biological phenomenon of hormonally induced sexual differentiation of the brain has been an empirical topic of study for over 50 years1 but much remains to be discovered in terms of both mechanism and functional impact. In the McCarthy lab we exploit the many advantages of the laboratory rat as our model because of its well-characterized neuroanatomy and behavioral repertoire which, as in humans, differs markedly in males and females. In particular we have drilled deeply into the mechanistic bedrock controlling male copulatory behavior with a focus on one brain region, the preoptic area (POA), which is often associated with the hypothalamus but is actually telencephalic in origin2. If this brain region is lesioned, males loose all interest in sex3 and if it is stimulated they loose all interest in anything but sex4. The importance of the POA to male reproduction cuts across species from newts to humans, but the mechanisms underlying its importance are only beginning to be understood in the rodent.
In the beginning, most studies justifiably focused on neurotransmitter production, release and binding, assuming that a change in behavior must be the result of changes in neural activity. But finding precisely what about neurons was different in males versus females proved frustrating and largely unproductive until attention turned to other factors. In particular we determined that prostaglandins, a normally inflammatory mediator, was both necessary and sufficient to masculinize the brain if elevated during a critical perinatal “sensitive period”5. Even more surprising, non-neuronal cells that constitute the brain’s immune system called microglia, are a principle source of the prostaglandins that masculinize the POA and male copulatory behavior6. Cell-to-cell communication that involves neurons, astrocytes and microglia helps to shape the synaptic profile of this brain region by increasing the density of excitatory spine synapses on dendrites of POA neurons.
In the course of our studies we noted that the sex difference in synaptic density, males having twice the number of spine synapses per unit of dendrite length as females, was stable across the lifespan despite the juvenile hiatus during which there are little to no gonadal steroids in circulation in either males or females. This led us to ponder the question of how this cellular memory was maintained, and to answer that we turned our attention to epigenetics.
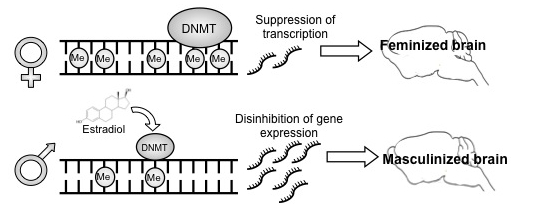
Steroid hormones are obvious candidates as genome modifiers because their transcription factor receptors are associated with co-factors that possess histone deacetylase activity. But to our surprise we found that rather than directly interacting with DNA or chromatin, steroids were instead (or more likely in addition to) decreasing the activity of a class of enzymes called DNA methyltransferases (DNMTs), lowering their ability to methylate DNA7. Reduction in the activity of these actively methylating enzymes results in demethylation of DNA through mechanisms not well understood. Regardless, the impact is an emancipation of genes that then direct masculinization of the POA resulting in male-typical synaptic patterning and copulatory behavior in adulthood. Moreover, if we allowed demethylation to occur outside of the sensitive period for sexual differentiation of the brain, females were still capable of being masculinized. The end of the sensitive period is operationally defined as the point at which giving masculinizing hormones to a female no longer has any effect on sexual differentiation of her brain. We confirmed that the sensitive period ends before day 10 after birth, and we also showed that hormones no longer reduce DNMT activity at this time. Thus the end of the sensitive period appears to be due to the loss of effect of steroids on DNMT activity. To our knowledge this is the first ever report of a mechanism for the end of the sensitive period.
The next big question of course is – what genes are demethylated in the bipotential brain leading to masculinization? Here we have a tale of woe as this is far harder to answer than it might at first seem. Our initial approach was to categorize gene expression profiles in males and females with and without DNMT inhibition using RNA-Seq. To our surprise there were a relatively small number of genes that showed overall expression differences in males and females, less than 100, and they were evenly distributed between males and females. We then looked to see what genes were turned on in females following demethylation. The expression of many genes was induced in the female POA follow DNMT inhibition, and importantly the majority that had been higher in males were now upregulated in females. These genes we considered our masculinization genes, which after eliminating unknowns was reduced to less than 10 candidate genes. The obvious next step was to characterize methylation of CpG islands found in the promoters of each gene. We chose to use the highly quantitative approach of 454 sequencing. However, there were several challenges inherent to this technique: its only good for amplicons of ~300-600bp, and so for large CpG islands several amplicons are required, meaning multiple pairs of primers per promoter. The book keeping alone is a nightmare and determining CpG methylation requires bisulfite conversion from a large number of biological replicates (in our case three groups with n’s of 8-10 per group). Thus the potential sources of error were insomnia inducing. But despite all this, with all quality controls in place, we found absolutely no meaningful sex differences in CpG methylation in any of our candidate genes. Nothing. At first this was very hard to believe, much less accept, but in retrospect, it makes sense.
The first clue that we should not have expected to detect differences in these candidate genes was hiding in our own data. The observation that DNMT activity is higher in the neonatal female POA was matched by global methylation levels of DNA extracted from the POA, such that females had twice the amount of methylation as males. Twice the amount of methylation on the entire genome should not reduce down to differences in the promoters of a handful of genes. Either our measure of DNA methylation was flawed, or most of the action was outside of the promoters. Turns out both are at least partially true.
Extraordinary claims require extraordinary evidence – a quote attributed to Carl Sagan and used by the dreaded Reviewer #3 to insist that if we were going to claim that females had double the DNA methylation in the POA as males we needed better proof than an antibody based colormetric assay kit. There are only a few roads to complete methylome analysis and most of them are full of potholes. The only superhighway is Genome Wide Bisulfite Sequencing (GWBS) but the tolls are very high and the data generated could circle the globe many times over. So what if we took the on ramp but crashed and burned, finding no sex differences in DNA methylation? These are the kinds of decisions one should not contemplate for long or you will talk yourself out of it. So we did it; collecting and bisulfite converting the DNA from the POA of newborn males, females and females masculinized by hormone treatment two days earlier, with 3 biological replicates per group, the industry standard. We shipped our samples off for sequencing and waited. It was agony until the data arrived by UPS, as the files were too large to send electronically, and then it was shear terror. Fortunately, one of us (BN) had gained sufficient bioinformatics experience to conduct an initial analysis and found that indeed, females do have twice the level of DNA methylation as males, but only at sites that are very highly methylated, not across the genome. In fact females also had more sites that were entirely unmethylated compared to males and masculinized females, suggesting tight epigenomic regulation in females. Analyses of where in the genome differences in methylation status between males and females can be found showed that most differences are in the intragenic regions, but of course this is where most methylation is found and so the significance of this remains unclear at the moment. Relatively few sex differences were found within CpG islands or shores, consistent with our failure to find any differences in the promoters of our candidate genes in our earlier attempts at 454 bisulfite sequencing.
The journey to publishing this paper had begun over two years earlier and we were anxious to put it to rest, submitting the final draft with the bare minimum of analyses of the GWBS data. But there is a treasure trove of information; we are anticipating more surprises reside therein. These will surely provide insight into the proximate mechanisms establishing and maintaining sex differences in this brain region, but will not answer the big picture question of why the brains of males and females evolved this way. At this point we can only speculate, and our speculations fall along two lines of thinking. The first is the process of canalization, which Waddington proposed as the function of epigenetic modifications and a process since considered widely in the context of evolution and the robustness of species in the face of challenge8. The robustness of sex differences in neuroanatomical endpoints (NOT behavior) is reminiscent of canalization and the marked differences in epigenetic marks we have observed may be a mediating factor. Second is a more tautological explanation based on the notion that the reproductive strategy of females requires close guarding of precious and limited gametes while males have a continuous and plentiful supply that they are eager to share. If females were to begin to play like males the consequences would be costly if not disastrous, therefore they actively suppress the gene profile that if activated, leads to masculinization of brain and therefore behavior.
More information on the McCarthy lab here.
1. PHOENIX CH, GOY RW, GERALL AA, & YOUNG WC (1959). Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology, 65, 369-82 PMID: 14432658
2. Puelles, L., Harrison, M., Paxinos, G., & Watson, C. (2013). A developmental ontology for the mammalian brain based on the prosomeric model Trends in Neurosciences, 36 (10), 570-578 DOI: 10.1016/j.tins.2013.06.004
3. Heimer, L., & Larsson, K. (1967). Impairment of mating behavior in male rats following lesions in the preoptic-anterior hypothalamic continuum Brain Research, 3 (3), 248-263 DOI: 10.1016/0006-8993(67)90076-5
4. Malsbury, C. (1971). Facilitation of male rat copulatory behavior by electrical stimulation of the medial preoptic area Physiology & Behavior, 7 (6), 797-805 DOI: 10.1016/0031-9384(71)90042-4
5. Amateau, S., & McCarthy, M. (2004). Induction of PGE2 by estradiol mediates developmental masculinization of sex behavior Nature Neuroscience, 7 (6), 643-650 DOI: 10.1038/nn1254
6. Lenz, K., Nugent, B., Haliyur, R., & McCarthy, M. (2013). Microglia Are Essential to Masculinization of Brain and Behavior Journal of Neuroscience, 33 (7), 2761-2772 DOI: 10.1523/JNEUROSCI.1268-12.2013
7. Nugent, B., Wright, C., Shetty, A., Hodes, G., Lenz, K., Mahurkar, A., Russo, S., Devine, S., & McCarthy, M. (2015). Brain feminization requires active repression of masculinization via DNA methylation Nature Neuroscience, 18 (5), 690-697 DOI: 10.1038/nn.3988
8. WADDINGTON, C. (1942). Canalization of Development and the Inheritance of Acquired Characters Nature, 150 (3811), 563-565 DOI: 10.1038/150563a0
 (3 votes)
(3 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.