The pattern of research
Posted by aaron_savage, on 2 April 2019
The correct patterning of embryonic tissues is essential for normal development. Aberrant patterning can lead to developmental abnormalities and pathogenic defects. Therefore, studying developmental patterning is important to better understand disease. The zebrafish embryo is a fantastic model for studying patterning during development owing to its optical clarity, small size and large clutch number. When coupled with dynamic transgenic lines, the picture of what occurs in the cell during these processes is starting to emerge.
Understanding how the ubiquitous second messenger, the calcium ion, regulates cellular physiology during development has become an important question in biology. This is where my story begins. I arrived in Sheffield after obtaining a PhD position, eager for what lay ahead. The project was exciting; using new transgenic lines and cutting-edge microscopy to study the function of a poorly understood gene, tmem33, which we hypothesised would regulate calcium signalling within the developing vasculature. The preliminary morpholino knockdown data generated in my host lab before I started my PhD hinted at a vast cache of riches waiting to be uncovered. This data suggested a role for tmem33 in both vascular and kidney development. I was given some new toys to play with, a light sheet microscope and an endothelial-specific calcium reporter line. My first experiment in the lab was to analyse the mutant zebrafish line generated by my supervisor around which my PhD was supposed to focus. It was, of course, completely normal. No vascular defects here. It looked like my project was dead in the water before it had begun. My next few experiments yielded additional dead ends – analysing 34 TRP channels by in situ hybridisation, looking for specific vascular enrichment (spoiler alert – there wasn’t any).
Thankfully, science moves very quickly. My original project went from dead, to very dead as several papers at the time heavily criticised morpholino-based approaches (Kok et al, 2014; Schulte-Merker and Stainier, 2014; Stainier 2017; Robu, 2007) to back alive within the space of about six months. A single paper (Rossi et al, 2015) brought hopes for a revived project. In a series of elegant experiments, the authors described a situation where knockdown of the egfl7 gene by morpholino induced a robust vascular phenotype, but when this gene was mutated using genome editing the phenotype was absent. Interestingly, the authors showed that the egfl7 mutant displayed nonsense mediated decay of egfl7 transcripts and that when egfl7 morpholinos were injected into egfl7 mutants, the mutants were protected against the effects of the morpholino. The synthesis of these data was that there existed genetic compensation in the mutants but not in the morphants.
Finally, the stroke of luck I needed, our tmem33 mutants also showed nonsense mediated decay, so I set about injecting tmem33 morpholinos into our tmem33 mutants and I found strikingly similar results. This suggested that the reason the tmem33 mutants displayed no phenotype was because they displayed a kind of genetic protection, likely via a genetic compensation mechanism. I now had the beginnings of a successful project, a year in to my PhD. Rossi et. al used a new technology to address their issues, CRISPR interference (CRISPRi) – a modified version of the CRISPR/Cas9 system using an inactivated form of Cas9 (dCas9). The authors were able to reproduce the same phenotype they observed via morpholino knockdown using CRISPRi. I applied CRISPRi to knock down tmem33 and was able to reproduce our morpholino knockdown data of tmem33.
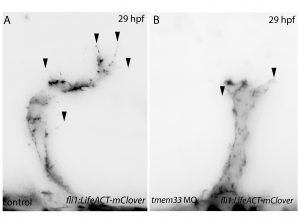
Next, inspired by the conditional CRISPR approaches described in Ablain et al (2015), I sought to conditionally knock down tmem33 in endothelial cells by driving dCas9 expression under the control of the endothelial-specific fli1a promoter. Preliminary experiments using transient conditional knockdown were promising – only endothelial cells showed a phenotype! The next step was to generate a transgenic line which stably expressed dCas9 in endothelial cells.
Throughout this process, I had been studying tmem33 function with regards to endothelial cell physiology during angiogenesis. Since it was known that tmem33 functioned within the endoplasmic reticulum, using a calcium reporter line I began to test whether tmem33 knockdown altered endothelial calcium signalling. I found that tmem33 knockdown reduced observable endothelial calcium oscillations, which coincided with reduced endothelial cell migration. This was validated by both morpholinos and CRISPRi. Furthermore, I began using both knockdown approaches to position the tmem33 within the hierarchy of developmental angiogenic signalling. I found that tmem33 functions downstream of VEGF signalling but upstream of Notch and ERK signalling, identifying an essential function for calcium oscillations in mediating the response to Vascular Endothelial Growth Factor (VEGF) and inducing downstream signalling pathways essential during angiogenesis.
Calcium signalling during endothelial development.
Throughout my PhD, I’ve learned how the zebrafish can be a powerful tool for understanding basic biology and how a PhD is not a linear endeavour. You have to follow the research (and the results of others) and see where it takes you. I’ve also found that phenotypes (or rather, the lack of them) can be hard to interpret. If you think about it, nothing would be alive today if there weren’t contingency plans built into the genome. Unexpected negative results (I’m looking at you again, non-phenotypic mutants) are not worthless. In fact, they’re probably more interesting.
References
Kok, Fatma O., et al. “Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish.” Developmental cell 32.1 (2015): 97-108.
Robu, Mara E., et al. “p53 activation by knockdown technologies.” PLoS genetics 3.5 (2007): e78.
Rossi, Andrea, et al. “Genetic compensation induced by deleterious mutations but not gene knockdowns.” Nature 524.7564 (2015): 230.
Savage, Aaron M., et al. “tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis.” Nature communications 10.1 (2019): 732.
Schulte-Merker, Stefan, and Didier YR Stainier. “Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology.” Development 141.16 (2014): 3103-3104.
Stainier, Didier YR, et al. “Guidelines for morpholino use in zebrafish.” PLoS genetics 13.10 (2017): e1007000.
 (3 votes)
(3 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.