The dynamics of chromatin when life begins
Posted by jingyi, on 20 July 2016
Fertilization marks the start of life. This is followed by highly coordinated epigenetic reprogramming that allows protamine-histone exchange, zygotic genome activation, and the generation of a totipotent embryo. However, the true state of chromatin at the level of DNA during this crucial period is a long-standing mystery.
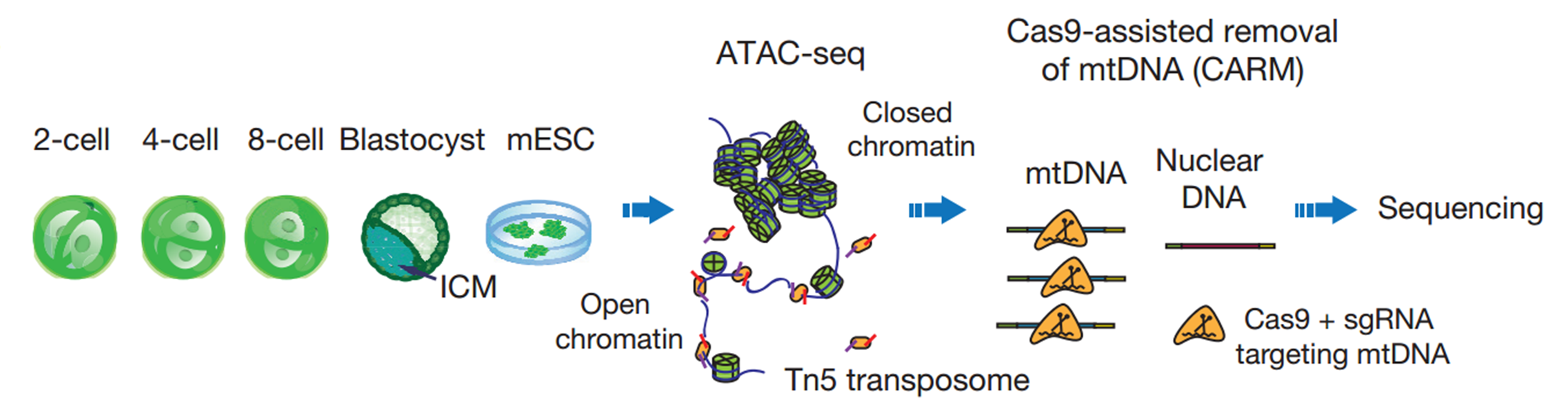
Our lab is dedicated to understanding epigenetic reprogramming in early development. But for a long time, we were struggling to find the right tools. When I first heard about ATAC-seq developed by the Greenleaf and Chang groups in Stanford, I was very excited as this is exactly the approach that we were looking for. I brought this up to my PI and we quickly decided to give it a try. We started by testing it in various numbers of mESCs, using either crosslinked or native chromatin from either frozen or fresh samples as well as different kinds of detergent in lysis buffer. In our hands, it appeared that ATAC-seq worked best on native chromatin of fresh samples from 100 to 1,000 cells with 0.15% to 0.5% NP-40 in lysis buffer.
We then began to apply ATAC-seq to mouse early embryos. Working together with another graduate student Bo Huang who has extensive experience in mouse embryos, we collected the early 2-cell, 2-cell, 4-cell, and 8-cell embryos, as well as ICMs isolated from blastocysts. However, we were surprised to see that the sequencing libraries were strongly contaminated by mitochondrial DNA, which occupied up to 99% of the reads. This is mainly due to the large quantities of mitochondria inherited from oocytes. My colleagues and I then spent almost a year to test several approaches to reduce mitochondrial contaminations. Most methods we tried did not meet our expectation, until we developed CARM, the Cas9-Assisted Removal of mitochondrial DNA. The 114 sgRNA evenly targeting 16kbp mitochondrion genome is designed to introduce the cutting event on mitochondrion DNA fragment in ATAC-seq library, so that these reads could not be sequenced due to the lack of sequencing adaptor on the both end of the fragments. We were very happy to see that CARM significantly reduced the about 70% of mitochondrial DNA at that time. Developing a new method is never easy, but the support from my mentor enabled me to proceed without any hesitation. He always encourages us to take the challenge to solve the problem that matters.
This story is now published on Nature as an article in June 30 (Wu & Huang et al., Nature 534 (652-657)). There are several interesting discoveries that we have made in this study.
Firstly, we found that unlike asymmetric DNA methylomes between the two alleles throughout preimplantation, accessible chromatin landscapes already become comparable between the two parental alleles after the 2-cell stage. Secondly, the open chromatin in early embryos occurs not only at the transcription start sites (TSSs) but also at the transcription end sites (TESs) of active genes, indicating a unique and unknown regulation mechanism in early development. Thirdly, we identified putative enhancers that are active in early development, through which we derived candidate transcription factors that may regulate early development. We validated the roles of two such regulators, Gata4 and Nr5a2, in lineage-specific transcription programs in preimplantation development. Finally, in early 2-cell embryos where major ZGA has not started, we found many large open chromatin domains (up to 110kb) over regions showing repeat-driven promiscuous transcription. These data support an unusually permissive chromatin state at this stage.
Taken together, our findings unveiled highly dynamic spatiotemporal configuration of chromatin states in early mammalian development. However, we believe this is just the beginning. Future investigations are warranted to fully unlock the mechanisms and the functions of these unique regulatory modes. As a graduate student, this was truly an exciting journey for me. The happiest thing is to make discoveries in a world full of unknowns.
 (4 votes)
(4 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.