An interview with Jenny Nichols
Posted by the Node, on 17 August 2017
This interview by Aidan Maartens originally appeared in Development, Volume 143, Issue 16.
Jennifer Nichols is a Principal Investigator at the Cambridge Stem Cell Institute and Department of Physiology, Development and Neuroscience at the University of Cambridge, UK. Her lab works on lineage segregation and the establishment of pluripotency in the mammalian embryo. In 2017 she was awarded the British Society for Developmental Biology’s Cheryll Tickle Medal, given to mid-career female scientists with outstanding achievements in developmental biology. We met Jenny in her Cambridge office to talk about pluripotency in vitroand in vivo, the importance of collaboration in her career path, and what playing a musical instrument has in common with research.

This year you were awarded the Cheryll Tickle Medal. What does the award mean to you?
Well it really meant a huge amount to me – the nominations come voluntarily from our peers, and then it’s voted on by people on the committee, so I was just really touched by that. And I hadn’t really thought of myself at all as someone who would receive medals.
Let’s start at the beginning: what got you into science in the first place?
My father was a naturalist interested in marine biology, and so when we went to the beach he’d be fishing around in rock pools and I’d tag along with him. So I was kind of brought up with biology that way – I always loved animals and finding things under rocks. I then went through education knowing that I wanted to do some kind of research: I quite liked physics and maths in the early days, but I could just see myself doing biology because I’d have the chance to work with animals.
Your first paper, published in 1984 in the Journal of Experimental Embryology and Morphology (the forerunner of Development), described the isolation and culturing of inner cell masses (ICMs) from the mouse embryo. How did that work come about?
In the final year of my degree, I knew I wanted to do developmental biology, and I wrote to various developmental biologists, one of whom was Richard Gardner. He wrote back to say that although he didn’t have anything open at the moment, he was always glad to meet people who were interested in developmental biology. So I hitchhiked up to visit him, and talked to him about the kind of work I was doing at the time, and it was great – I really liked him and liked Oxford. And then a couple of months later he wrote to tell me that his postdoc, Ginny Papaioannou, was going to be leaving. This was in the good old days when there were positions and resources available in universities and one didn’t have to depend on grants, so when Ginny left there was space in the lab. Richard asked whether I was interested in becoming his research assistant, and I arrived in his lab in 1981.
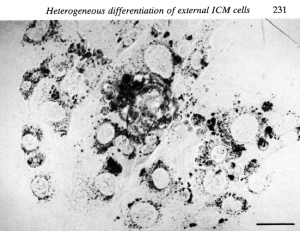
In terms of how the paper came about, in the 1950s and 1960s the work of Tarkowski and colleagues had suggested that the development of the trophectoderm was very much dependent upon position within the early embryo. By the time I joined Richard’s lab things had moved on a little bit – for instance, it became possible to isolate the ICM and culture it, and that’s what Richard and other people such as Alan Handyside had been doing. This work led to the notion that, in addition to position, timing was crucial too: isolated ICMs from very early blastocysts could regenerate the trophectoderm, whereas those from later embryos gave rise only to epiblast and primitive endoderm. When you cultured ICMs from an intermediate stage, you could get both trophectoderm and primitive endoderm, at least by morphology, and that first paper focussed on the question of whether the outer layer of the ICM was a mosaic of the two cell types. We never really got to the bottom of what it all meant at the time, but the most important aspect of that work for my career was that I became an expert in dissecting embryos and culturing ICMs. And because I’d inherited the lab space from Ginny, there was a micromanipulation rig and a dissecting microscope for me: I had all this kit, and the mice were provided by the institution, so I had the opportunity while I was there to develop hand skills.
In Gardner’s lab you worked alongside Rosa Beddington: I understand she was a great influence on you?
Rosa was just finishing her PhD as I joined. She was incredibly clever and very good at manipulating embryos, and wouldn’t take no for an answer – if she wanted to do something, she’d find a way, and that made you think that if Rosa could do it, perhaps you could do it too.

For me, I think the most important thing she did was to invite me to help on the Cold Spring Harbor course ‘Molecular Embryology of the Mouse’. I started there as a teaching assistant just before I left Oxford in 1990. At the time I didn’t really have a great deal of confidence, being overshadowed by all the brilliant people in Oxford, and being a teaching assistant gave me something that I could do usefully. I also got to meet all of these amazing embryologists, and of course to mix with the participants. That gave me a big confidence boost and got me in contact with people in the field.
You moved from Oxford to Edinburgh with Austin Smith, getting your PhD in 1995 and then working as a postdoc. How did you come to meet Austin and, in the early days of his lab, what were the key questions you were aiming to answer?
When I was with Richard, Austin had come to work with John Heath across the road – he’d been interested in embryonic stem cells (ESCs), and his PhD had been devoted to finding what it was in the feeder layer on which mouse ESCs were grown that allowed them to self-renew and propagate. Being a biochemist, Austin wanted to understand what signalling pathways were involved, and then figure out how to make the process more efficient and relate it better to the embryo. John Heath introduced me to Austin and suggested that we might want to work together, and it took off from there. Being in Richard’s lab, everyone was a brilliant embryologist, but working with Austin – who was a biochemist and didn’t do embryo work – provided me with a niche. Then, when Austin got the job in the Centre for Genome Research in Edinburgh – another core-funded venture – he asked if I would go with him.

During his postdoc Austin had done HPLC to analyse what components in the medium secreted from the feeder layer promoted ESC renewal and found the active agent, which he called differentiation inhibitory activity (DIA) and turned out to be leukaemia inhibitory factor (LIF). So then the question was: if LIF is able to maintain ESCs in a self-renewing state in vitro, does it have any relevance in the embryo? When I started working with him, we wanted to look at the cells in culture, but also to understand how precursor ESCs in mouse embryos can self-renew. During my PhD I did in situ hybridisation on components of the LIF pathway in the embryo. In situ hybridisations had been going for a few years but were radioactive and quite laborious, and I spent a lot of time cutting sections of embryos from wax in preparation.
After many years in Austin’s lab you became an independent Principal Investigator (PI) in 2006 at the Cambridge Stem Cell Institute. How did you find the transition?
When Austin was planning to move down to Cambridge, I had a bit of uncertainty over whether to go with him, but decided in the end that it was the right move – we got on very well and made a very good partnership, and of course we still had so much left to discover about the system. It was an unusual transition to being a PI – all of a sudden, I was! But I hadn’t really thought about it; as far as I was concerned I worked with Austin. We did, by then, have joint grants, but my purpose at that time was to set up the transgenics facility. I didn’t have a fellowship but I did have a permanent job, and independence was a gradual transition from then, so I didn’t really notice any difference.
It seems that your research has been collaborative from the start. Does this reflect how you see science?
It’s how I see science going. In the old days people could get single-author papers, but I never really felt up to that, and so was always glad to collaborate. And I think that now it’s good advice that you collaborate – you need to have so many aspects to what you are doing and you can’t possibly be an expert in every one. The expectations for what is supposed to go into a paper are also very different from when I started. Plus, I enjoy collaborating and working with other people.
It’s good advice that you collaborate – you need to have so many aspects to what you are doing and you can’t possibly be an expert in every one
Since 2006 you have continued to work on lineage decisions and pluripotency in the early mouse embryo. What is your current understanding of how pluripotency arises, and the key open questions in the field?
The question of how the epiblast becomes pluripotent is of course fascinating in itself, but it’s also something you can get funding for. If I’d taken the other approach and focussed on primitive endoderm specification, even though it’s basically the same question, it’s much less easy to justify.
From research performed with a brilliant postdoc, Thorsten Boroviak, we’re pretty clear that the state of pluripotency in mice is acquired in the epiblast cells as a result of contact with the extracellular matrix that comes from the primitive endoderm cells as they are becoming specified. Thorsten found that the primitive endoderm cells are producing laminin and fibronectin at this stage, and that single cells from the ICM of earlier embryos would now make ESC colonies simply by the addition of the right matrix components. So it seems that interaction with factors secreted by neighbouring primitive endoderm cells is how pluripotency is established.
One of the key questions in the field is how cells start deciding which lineage to go down. Back when I started in Richard’s lab, the trophectoderm was known to form from the outside cells, and as the embryo was about to implant one could see the epiblast and the primitive endoderm, with the latter on the outside of the ICM. We assumed in those days that the cells that were exposed to the cavity would be the ones that would make the primitive endoderm – that it was solely a positional thing. So it was quite a breakthrough when Clare Chazaud and Janet Rossant, and then Berenika Plusa and Kat Hadjantonakis, showed that the primitive endoderm fate is specified in a salt-and-pepper distribution in the ICM. We still don’t really know how they start deciding which one to become, and the basis of this decision making is, I think, one of the key open questions.
How did working with human ESCs change the way that you thought about pluripotency?
Once we could derive human ESC lines – and it took quite a long time – it became clear that they are different from mouse ESCs. They make two-dimensional rather than small and dome-shaped colonies, and couldn’t be derived from single cells, suggesting that the population has to grow intact before passaging. They look different, express different molecular markers, require different factors in the culture and different ways of passaging. So the big question was what is the difference between the mouse and human ESC states? Then two groups, one headed by Roger Pedersen and the other by Ron McKay, derived what they called epi-stem cells (epiSCs) from the epiblast of later stage mouse embryos, using the culture conditions that had been successfully employed to make human ESCs. The logical explanation for the difference between mouse and human ESCs is, therefore, that during human ESC derivation the cells advance to the equivalent of a post-implantation state during culturing, and you recover cells in what we subsequently called the ‘primed’ pluripotent state.
So then the question was whether a condition exists in the human embryo or in other mammals similar to the mouse ESC state? EpiSCs can make pretty much every tissue you need, so would the embryo require a more naïve state? From my point of view, I was interested in the developmental biology. Others were also interested from a practical perspective, as mouse ESCs are so much easier to culture and you can do gene targeting more readily. When Takahashi and Yamanaka showed that one could reprogram differentiated cells to pluripotency – generating induced pluripotent stem cells (iPSCs) – this raised the possibility of reprogramming primed human lines to the naïve state. Many papers came out, incrementally addressing this question. Within the last few years two postdocs in Austin’s lab, Yasu Takashima and Ge Guo, found conditions in which many of the pluripotency markers in the mouse embryo were expressed differentially from the primed human cells, and crucially these were expressed in the human embryo.
There do seem to be quite a few paths to pluripotency across mammals. As far as I know, all mammals make a structure that looks like a blastocyst, but then what differs is what happens after that, how quickly it happens, and the pathways that are involved. For example, in the mouse primitive endoderm specification is entirely dependent on FGF signalling, but this is not the case in other species such as cattle or humans. Rodent embryos have quite a special way of developing that involves the formation of ‘egg cylinders’, unlike other mammalian species, and this structural difference might be key. Non-rodent embryos form a flat, polarised epithelium of the epiblast quite quickly, which might be significant in terms of the endurance of the naïve pluripotent state in the embryo.
In a time when iPSCs are being increasingly employed by developmental and stem cell biologists, what do mouse or human ESCs still have to offer us?
iPSCs have obvious uses and benefits, although of course it’s a given that if we hadn’t had ESCs then iPSCs wouldn’t have come about. But deriving iPSCs can be inefficient and take time, whereas if you want to derive an ESC line from a transgenic mouse line it is very easy from embryos, and the cells are pristine. For all these derivations, the acid test is whether they can make a chimera when put into an embryo, and whereas ESCs generally make good chimeras, iPSCs can be a bit fussier.
That is from the mouse perspective; from the human point of view, I have just established a collaboration with Wolf Reik where we are trying to derive human ESC lines clonally from embryos donated from IVF programmes. About 60% of these embryos are thought to be mosaic for aneuploidies, and a lot of those aneuploidies are likely to be chromosomal defects like trisomies. So they are actually potentially useful models for in vitro modelling of disease, and by being able to derive multiple clonal lines from an individual human embryo, one can powerfully compare a normal line with an abnormal line. This has the potential to be very useful and isn’t something we can do with iPSCs.
Don’t just follow fashions – you need a deep-rooted question that you wake up thinking about
Do you have any advice for young scientists?
Well, I said earlier that collaboration is important. In addition I think that, especially these days, you should find something that you are really interested in. You should be obsessed with your work. Don’t just follow fashions – you need a deep-rooted question that you wake up thinking about.
Finally, is there anything Development readers would be surprised to find out about you?
Several people probably know this but I am a keen oboe player and play in local orchestras. I think this is definitely connected to being a preimplantation mammalian developmental biologist – when we’re moving our embryos around, we use a pulled-out Pasteur pipette that we control with a mouth tube. And if you play a wind instrument, you don’t dribble! In playing musical instruments, you know you have to practise, and you know you’re not going to be brilliant straight away; learning how to dissect mouse embryos is similar – you can’t just follow a protocol and expect data immediately.
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.