Moving proteins within living embryos using light
Posted by Clare Buckley, on 24 February 2016
By Clare Buckley and Rachel Moore
One of the things that we find most challenging about working with whole vertebrate organisms is how we can tie ourselves in knots trying to unpick the function of a single component within such an intricate and interconnected web of proteins and signalling cascades. All too often knocking out (or down) a single gene produces a widespread phenotype, making it near impossible to differentiate between direct and indirect consequences. Either that, or redundancy within the system means that there is no phenotype at all. The recent morphant vs. mutant vs. CRISPR debate highlights this difficulty (Kok et al., 2015; Rossi et al., 2015; Schulte-Merker and Stainier, 2014).
The ‘chicken and egg’ conundrums of cell polarity
We were getting particularly frustrated with this problem while trying to work out how to test a feedback loop that we proposed following our finding of a novel mechanism of cell polarisation during zebrafish hindbrain lumen formation. Using live confocal imaging, we found that neural progenitor cells locate their centrosomes and assemble apical polarity complexes to whichever point they intersect the middle of the developing tissue, even if this is part-way along a cell length (Buckley et al., 2013).
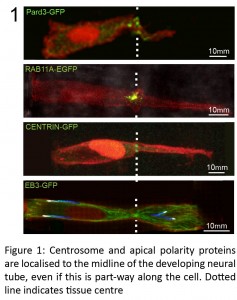
This initiation of apical polarisation part way along the length of a cell, rather than at a cell extremity, was unexpected and raised the question of how these cells ‘know’ where to initiate apical polarity establishment. We proposed a polarisation feedback loop by which initial Pard3 puncta specify centrosomal location, causing the centrosome to organise the microtubule cytoskeleton around the tissue midline, which is in turn necessary to reinforce Pard3 delivery to this point.
Whilst watching the localisation of individual proteins during morphogenesis often tells us a lot about their function, directly testing this feedback loop will require us to manipulate one or more of its components. Rather than abrogating the function of e.g. Pard3, a much more powerful approach in this case would be to physically MOVE one of the components of the proposed feedback loop, to see which other components followed it. In order to do this, we needed a technique that would allow us to target specific proteins and to move them subcellularly with high temporal and spatial resolution.
The phytochrome system
We were mulling over this problem when we attended a conference on polarity at the Royal Society, where Orion Weiner presented his lab’s fantastic work using the Phytochrome system to manipulate subcellular protein localisation in cultured cells and yeast (Levskaya et al., 2009; Toettcher et al., 2013; Yang et al., 2013). For example, Levskaya et al. (2009) neatly illustrated that they could control lamellipodia extension in mammalian cells by targeting Rho-GTPases to specific sites in the cell membrane. Originally found in Arabidopsis, the phytochrome system consists of three components: the phytochrome B protein (PHYB); a basic-helix-loop-helix transcription factor called phytochrome interaction factor (PIF; PIF3 or PIF6); and an external chromophore, phycocyanobilin (PCB). PHYB and PIF are induced to bind in the presence of far-red light and to disassociate under infrared light. This interaction is dependent on PHYB being bound to the chromophore PCB (Ni et al., 1999).
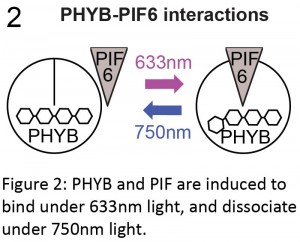
The phytochrome system therefore had all of the characteristics we were looking for: spatiotemporal control that was not only fine-scale, but also reversible. It also had the advantage of utilising wavelengths (far-red and infrared) that we don’t often use for imaging cells, so we would be able to label and image our cells using standard green fluorophores. If only we could use this system in whole vertebrates!
Luckily for us, Orion and his lab share our view that collaboration is one of the keys to good science and Clare was able to visit his lab at UCSF on an EMBO short term fellowship to develop the phytochrome system so that it could be used in live zebrafish embryos. After a few months of cloning, swearing (relatively quietly) at the confocal microscope, fantastic interactions with the lab and plenty of Anchor beer, it finally worked! (Science being science, this occurred at 2am after some slightly louder swearing and with the aid of another late-working postdoc who helped Clare’s sleep-deprived brain switch the microscope on properly.)
The following year, back in Jon Clarke’s lab at King’s College London, we were able to develop the Phytochrome system further and use it in combination with live imaging of the developing zebrafish hindbrain, which resulted in our paper describing this method (Buckley et al., 2016).
Rapid and reversible
During the resubmission of our paper, the Phytochrome system was used to demonstrate nuclear protein import in superficially located zebrafish cells using whole-embryo illumination (Beyer et al., 2015). However, it had not yet been used in multicellular organisms for fine-scale spatiotemporal or reversible control of protein localization, or for accessing cells deeper within the tissue. Following optimization of the PHYB protein and of PCB chromophore purification and delivery, the phytochrome system worked well in zebrafish embryos. We injected a combination of a membrane-bound PHYB-mCherry-CAAX and cytoplasmic PIF6-EGFP mRNA along with PCB and measured how quickly the binding was turned on and off by analysing the intensity of EGFP fluorescence in the cytoplasm compared to the membrane. We found that the PHYB-PIF interaction provided us with rapid temporal control. Using the 633 nm laser on our confocal (far-red light induces PHYB-PIF interaction), PIF6-EGFP was recruited from the cytoplasm to the membrane with a time constant of 6.5 seconds. Conversely, PIF6-EGFP was released from the membrane into the cytoplasm at a time constant of 46.9 seconds under infrared light (which causes PHYB and PIF to dissociate) from a standard light LCD light source fitted with an infrared filter. This cycle of turning the binding off and on could be repeated several times without bleaching. And, crucially, we were successfully able to recruit the PIF6-EGFP construct to specific regions of the plasma membrane by restricting the far-red light (turning binding on) to a small region-of-interest whilst simultaneously bathing the entire sample in infrared light (turning binding off).
Beginning to address our original question – can we manipulate polarity protein location?
Of course, our original aim was to manipulate the polarity proteins from our putative feedback loop. To do this, we made a Pard3-PIF6-EGFP construct and confirmed that it showed a similar localisation to endogenous Pard3. We were able to direct its subcellular localisation into small regions-of-interest in the thin, flat epithelial cells of the enveloping layer, as well as in neuroepithelial cells in the developing neural tube.
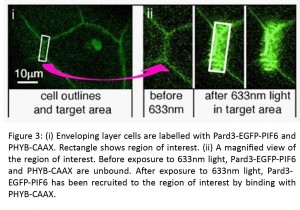
Meanwhile, another project of Clare’s resulted in the birth of her daughter in April! Rachel heroically took up the mantle and produced some fantastic data showing that the specific enrichment of Pard3-PIF6-EGFP also resulted in the recruitment of the binding partner protein Pard6-MCherry to the same area, illustrating that Pard3 maintains its activity despite our manipulations. This demonstration of the spatiotemporal control of Pard3 activity is very exciting for us and opens up many possible experimental avenues. For example, during neural tube development, the inheritance of Pard3 by one daughter cell following mitosis is associated with neuronal fate (Alexandre et al., 2010; Dong et al., 2012). As a proof-of-principle experiment, we used the phytochrome system to force the Pard3-PIF6-EGFP into one daughter cell during a mitotic division. We think that this is an excellent example of the powerful experiments that will be possible using the phytochrome system.
Future work
There are a couple of further modifications that we think will make the phytochrome system even more robust. One obvious step forward would be to develop fish lines that express various components of the phytochrome system – PCB, for example, or the membrane-bound PHYB-mCherry-CAAX, are two that would be particularly useful.
Another important fact to remember is that we are not labelling – and therefore not disturbing – the endogenous Pard3 protein. To perfect this system we will need to either remove the endogenous protein before expressing the modified, PIF6-bound protein, or else insert the PIF6 sequence into the appropriate location close to Pard3. New techniques such as CRISPR should make the latter relatively straightforward.
We’re really looking forward to applying the phytochrome system to other questions in our lab as well. We also hope that it will be a useful tool for the developmental biology community. We envisage that such a subtle manipulation of proteins could be used to address a variety of problems, and we hope that it will be able to be applied to other model organisms. As such, we would encourage anyone who would like to try it out to contact us for constructs or a chat, and we look forward to reading about more experiments utilising the phytochrome system for developmental biology and beyond.
Alexandre, P., Reugels, A.M., Barker, D., Blanc, E., and Clarke, J.D. (2010). Neurons derive from the more apical daughter in asymmetric divisions in the zebrafish neural tube. Nature neuroscience 13, 673-679.
Beyer, H.M., Juillot, S., Herbst, K., Samodelov, S.L., Muller, K., Schamel, W.W., Romer, W., Schafer, E., Nagy, F., Strahle, U., et al. (2015). Red Light-Regulated Reversible Nuclear Localization of Proteins in Mammalian Cells and Zebrafish. ACS synthetic biology 4, 951-958.
Buckley, Clare E., Moore, Rachel E., Reade, A., Goldberg, Anna R., Weiner, Orion D., and Clarke, Jonathan D.W. (2016). Reversible Optogenetic Control of Subcellular Protein Localization in a Live Vertebrate Embryo. Developmental Cell 36, 117-126.
Buckley, C.E., Ren, X., Ward, L.C., Girdler, G.C., Araya, C., Green, M.J., Clark, B.S., Link, B.A., and Clarke, J.D. (2013). Mirror-symmetric microtubule assembly and cell interactions drive lumen formation in the zebrafish neural rod. The EMBO journal 32, 30-44.
Dong, Z., Yang, N., Yeo, S.Y., Chitnis, A., and Guo, S. (2012). Intralineage directional Notch signaling regulates self-renewal and differentiation of asymmetrically dividing radial glia. Neuron 74, 65-78.
Kok, Fatma O., Shin, M., Ni, C.-W., Gupta, A., Grosse, Ann S., van Impel, A., Kirchmaier, Bettina C., Peterson-Maduro, J., Kourkoulis, G., Male, I., et al. (2015). Reverse Genetic Screening Reveals Poor Correlation between Morpholino-Induced and Mutant Phenotypes in Zebrafish. Developmental Cell 32, 97-108.
Levskaya, A., Weiner, O.D., Lim, W.A., and Voigt, C.A. (2009). Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997-1001.
Ni, M., Tepperman, J.M., and Quail, P.H. (1999). Binding of phytochrome B to its nuclear signalling partner PIF3 is reversibly induced by light. Nature 400, 781-784.
Rossi, A., Kontarakis, Z., Gerri, C., Nolte, H., Holper, S., Kruger, M., and Stainier, D.Y.R. (2015). Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230-233.
Schulte-Merker, S., and Stainier, D.Y.R. (2014). Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology. Development 141, 3103-3104.
Toettcher, J.E., Weiner, O.D., and Lim, W.A. (2013). Using optogenetics to interrogate the dynamic control of signal transmission by the Ras/Erk module. Cell 155, 1422-1434.
Yang, X., Jost, A.P., Weiner, O.D., and Tang, C. (2013). A light-inducible organelle-targeting system for dynamically activating and inactivating signaling in budding yeast. Molecular biology of the cell 24, 2419-2430.
 (4 votes)
(4 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.