Two birds with one stone: CTCF control of dynamic gene expression during heart development.
Posted by mgv, on 9 October 2017
CTCF binds to chromatin and is thought of as an architectural protein in the genome. If the genome were a text, CTCF would act like the punctuation marks, so that words are grouped together becoming meaningful sentences.
When I started my PhD, the Manzanares lab had been fruitfully collaborating with that of Jose Luis Gómez-Skarmeta at the CABD in Seville for several years in different projects. In one of them, they were studying IrxA gene regulation in the developing embryo, and previous results they had generated suggested that CTCF had a role in the regulation of this cluster. This is a fascinating gene cluster, duplicated during vertebrate evolution, which comprises only three homeobox-containing genes spread over more than one megabase of DNA. It has three genes, the two first (Irx1 and Irx2) showing nearly identical expression patterns, while the third gene, Irx4, shows distinct prominent expression in the heart.
The ideal experiment we first thought of was to delete a particular CTCF binding site. Another option was to prevent CTCF protein from binding there. This could be achieved by using a conditional mouse mutant allele, where Ctcf is flanked by two loxP sites (1), which allows the specific deletion of this gene, in our case using a Cre-driver specific for the developing heart. Other groups had already used this Ctcf floxed line and conditionally deleted the gene in several contexts, such as the developing limb or T-cells, finding that CTFC had roles in processes such as apoptosis or proliferation.
Although we initially aimed to study the function of a particular CTCF binding site, I started characterizing the phenotype derived from the lack of CTCF in the developing heart. Quickly, this experiment acquired life of its own, when we saw that deleting CTCF during heart development led to embryonic death. This highlighted the importance of CTCF in the process, and thus we decided to focus on the overall function of CTCF in the developing heart.
In order to obtain a global view of the effects on gene regulation by Ctcf-deletion we carried out RNA sequencing of control hearts and of other two scenarios: one or two copies of Ctcf deleted. We found that only one copy of Ctcf is enough for correct gene expression and normal heart development (mice reach adulthood with no problems) so we focused on the comparison of gene expression between wild-type and Ctcf homozygous mutant embryonic hearts.
When we carried out functional annotation of the list of approximately two thousand genes with significant changes in expression between wild-types and mutants, the Gene Ontology (GO) term “heart development” stood out in all different GO tools we used. What also caught our attention was that the “heart development” and related GO terms were only present in the down-regulated gene set. To corroborate this finding we analyzed the expression pattern of some of the genes belonging to the “heart development” GO term (such as the master cardiac regulator Nkx2-5) by in situ hybridization, and we could observe clear downregulation of all genes examined. This led us to think that CTCF controlled heart development by directly regulating those genes critical for embryonic patterning.
Our next question was pretty straightforward: what is the relationship between CTCF binding to the genome and our set of differentially expressed genes in the Ctcf knockout? Taking advantage of the previously described genome-wide distribution of enhancers and CTCF binding generated by ChIP-seq in the mouse heart by Bing Ren’s lab, we tried to tackle this question. The analysis showed that both up and down-regulated genes were closer to a CTCF binding site than expected, but that only down-regulated genes were closer to a heart enhancer. This observation suggested that CTCF is acting to bring together gene promoters and developmental enhancers to achieve proper expression in this developing system.
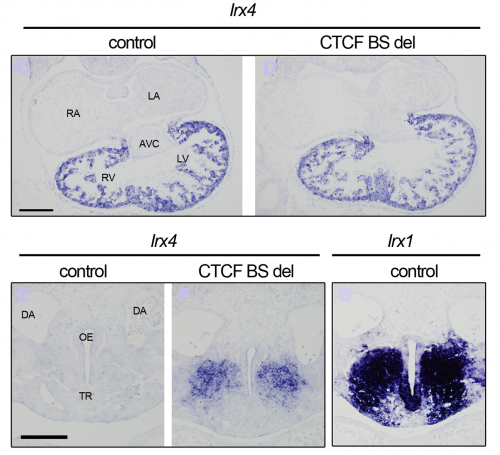
Following this last idea, we wanted to study in more detail if the alteration in transcription of selected genes in the absence of CTCF was due to changes in the 3D chromatin structure. When we were developing this work, chromatin conformation capture techniques (3C) and its derivatives were the way to go to study 3D chromatin structure, so we decided to use this emerging technology to address the issues we had in hand. At this point, two questions came up. First, how do we choose interesting candidates for further studies among 2000 genes. And second, which 3C-derived technique should we use? For the first, we chose Irx4, as it is one of the members of the IrxA gene cluster, on which the Manzanares and Gómez-Skarmeta labs had been already working, and it has an important role in ventricle identity (2,3). Most importantly, we had seen that it was strongly downregulated in our RNA-seq data, had predicted heart enhancers and ChIP-seq CTCF binding sites in its vicinity, and in situ hybridization confirmed its down-regulation in mutant hearts. Regarding the 3C-derived technique to use, we chose 4C-seq because we knew very little about Irx4 regulatory landscape and in this way we could address all the interactions occurring from specific regions in the locus, as this is a “one-versus-all” approach.
Standard 4C-seq protocols are supposed to use 1×107 cells, and this was a challenge given that we planned to carry out the analysis using E10.5 embryonic hearts, that are composed of roughly 30-40 thousand cells. For example, in a previous work showing p300 ChIP-seq in E11.5 embryonic hearts, 270 hearts were used (4). 270!! Obtaining wildtype hearts for setting up the technique was relatively straightforward, but Ctcf heart-specific mutants would only be one fourth of the litters. We managed to obtain reproducible 4Cs using pools of 45-60 for each genotype and replicate, but each pool took 2-3 months and long hours on the dissecting scope to collect. Nevertheless, the help of Claudio Badía-Careaga made this enormous effort, and the project in general, much more bearable.
Our 4C-seq showed that the Irx4 promoter interacted with surrounding CTCF binding sites. One of them was located between Irx1/Irx2 and Irx4, and therefore a prime candidate to be mediating the differences in expression of these two IrxA genes in the developing heart. To see if this site was important for regulating Irx4 in vivo, we deleted it with the new CRISPR-Cas9 system that Isabel Rollan in our lab had been working to set up. We could observe a subtle decrease of Irx4 expression levels in the heart when we compared the deletion mutant to controls. But, we were so focused in heart expression that we almost miss the fact that we had Irx4 ectopic expression. And it was in an Irx1 expression territory!! Thus, this CTCF binding site is crucial for proper Irx4 expression, both by aiding in its regulation by heart specific enhancers, and also by prevents its ectopic expression in territories where Irx1/Irx2 are normally expressed, possibly by not allowing other tissue-specific enhancers to activate the gene.
We were very pleased with these results, but we struggled for a while in publishing our data. We presented the work at several meetings and in one of them, somebody asked what happened with the upregulated genes, what are those? And why are there no heart development genes among them? Sure enough, we had looked at them but up to then merely described the functional enrichments. Therefore, we went back and re-checked the GO terms associated with up-regulated genes in Ctcf mutant hearts. Two categories stood out by the number of genes up-regulated: translation and mitochondria. The great majority of ribosomal protein-coding genes from both small and large subunits were among this subset, and more than 300 genes labelled with mitochondrial GO terms were up-regulated. When cardiomyocytes mature, they require two things in abundance: protein to build sarcomeres and energy to contract. Therefore, loss of Ctcf appeared to push cardiomyocytes towards maturation.
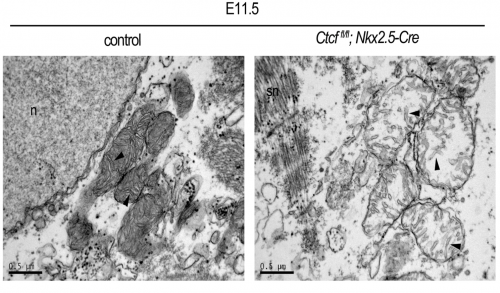
Looking at the mitochondrial genes differentially expressed in our RNA-seq, we found both functional and structural genes. We wanted to see if this upregulation led to more mitochondria and if these were functional. This was a new territory for us, and we were extremely lucky that in our same institute we had colleagues that are world- experts in mitochondrial biology. Ana Victoria Lechuga-Vieco, Rocio Nieto-Arellano and Jose Antonio Enriquez guided us through the analysis of the mitochondrial phenotype in our mutants. The RNAseq analysis showed an increase in several OXPHOS (oxidative phosphorylation) system components. We also saw this increase at protein level. However, the assembly of some OXPHOS super-complexes was impaired. One of my favorites results were the pictures Ana Lechuga-Vieco took with TEM (transmission electron microscopy). Not only they were absolutely beautiful and looked like a biology text book, but they showed that loss of CTCF lead to immature and sick-looking mitochondria, and that the sarcomeres assembled earlier than expected in the Ctcf mutant hearts. The 6 month-long wait for the TEM to be available had been worth it! Therefore it appears that despite increased transcription of maturation genes, proper assembly of cellular components does not follow leading to non-viable cardiac cells.
We concluded that CTCF was controlling two transcriptional programs in opposite directions, and this dynamic was necessary for proper formation of the heart. It is curious how the attempt to close a story opened a new one. And also, how a fresh look at your data can change the course of the story.

This post was written by Melisa Gómez-Velázquez and Miguel Manzanares
PLOS Genetics paper: CTCF counter-regulates cardiomyocyte development and maturation programs in the embryonic heart.
Bibliography
- Helen Heath, Claudia Ribeiro de Almeida, Frank Sleutels, Gemma Dingjan, Suzanne van de Nobelen, Iris Jonkers, Kam-Wing Ling, Joost Gribnau, Rainer Renkawitz, Frank Grosveld, Rudi W Hendriks, and Niels Galjart. CTCF regulates cell cycle progression of αβ T cells in the thymus. EMBO J. 2008 Nov 5; 27(21): 2839–2850. doi: 10.1038/emboj.2008.214. Epub 2008 Oct 16.
- Bruneau BG, Bao ZZ, Fatkin D, Xavier-Neto J, Georgakopoulos D, Maguire CT, et al. Cardiomyopathy in Irx4-deficient mice is preceded by abnormal ventricular gene expression. Mol Cell Biol. 2001;21(5):1730–6. doi: 10.1128/MCB.21.5.1730-1736.2001.
- Bao ZZ, Bruneau BG, Seidman JG, Seidman CE, Cepko CL. Regulation of chamber-specific gene expression in the developing heart by Irx4. Science. 1999;283(5405):1161–4.
- Matthew J. Blow, David J. McCulley, Zirong Li, Tao Zhang, Jennifer A. Akiyama, Amy Holt, Ingrid Plajzer-Frick, Malak Shoukry, Crystal Wright, Feng Chen, Veena Afzal, James Bristow, Bing Ren, Brian L. Black, Edward M. Rubin, Axel Visel, and Len A. Pennacchio. ChIP-seq Identification of Weakly Conserved Heart Enhancers. Nat Genet. 2010 Sep; 42(9): 806–810. Published online 2010 Aug 22. doi: 10.1038/ng.650.
 (2 votes)
(2 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.