Using human development to improve myogenesis from human pluripotent stem cells
Posted by Michael Hicks, on 21 February 2018
A discussion of our recent paper: Hicks et al., ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nature Cell Biology, January 1 2018
Approaching the problem of muscle disease with cell replacement therapy
We will all be affected by a muscle wasting disease in our lifetime. The ability to transplant a stem cell into skeletal muscle that can self-renew to make more of itself, and respond to injuries as they arise, could provide a continuous source of cells to replace damaged muscle tissue throughout our life span.
Stem cell replacement is especially imperative to the largest childhood muscle disorder Duchenne Muscular Dystrophy, where a genetic mutation in muscle cells results in muscle death and progressive weakness for about 20,000 new cases each year worldwide. The muscle stem cells of Duchenne patients also contain the genetic mutation rendering them ineffective at repairing the muscle as it dies.
Human pluripotent stem cells (hPSCs) have the potential to differentiate into any cell type making them attractive candidates for drug screening and cell replacement therapies. Indeed, hPSCs are already being used in the clinical setting and in preclinical models for diseases ranging from macular degeneration to cardiac myopathy, diabetes, and Parkinson’s disease.
Using induced pluripotent stem cells taken from Duchenne patients, our team at UCLA has devised a strategy to permanently correct the genetic mutation in Duchenne cells with CRISPR/Cas9 (Young et al., 2016). Our current challenge is delivering the corrected cells back to the affected patients. Correction of patient-derived induced pluripotent stem cells ex vivo and subsequent differentiation to human muscle stem cells offers one potential route for restoration of dystrophin after transplantation.
Pluripotent stem cells must mature through stages of development similar to natural cells to function like in vivo cell counterparts
When we first started this project, the only way to make skeletal muscle from human pluripotent stem cells was using virus to overexpress muscle transcription factors, which lacks precision in truly recapitulating the cells created from human development. For example, during regeneration a muscle stem cell must be able to simultaneously make more of itself and differentiate to make new muscle fibers. Forced viral overexpression of genes may limit proper stem cell dynamics.
Directed differentiation is a process that follows developmental cues to allow hPSCs to form any cell lineage. It has been well established that for many cell lineages, including cardiac muscle cells, the progeny made from directed differentiation of hPSCs are immature and do not function as well as adult cells. While there are a number of recent papers that use directed differentiation to produce muscle cells from hPSCs, no papers have benchmarked the cells being made to those derived from human muscle tissue. In this study we set out to follow human development of muscle more closely to create a muscle cell similar to those naturally found within the body as a means to obtain a cell for replacement therapy.
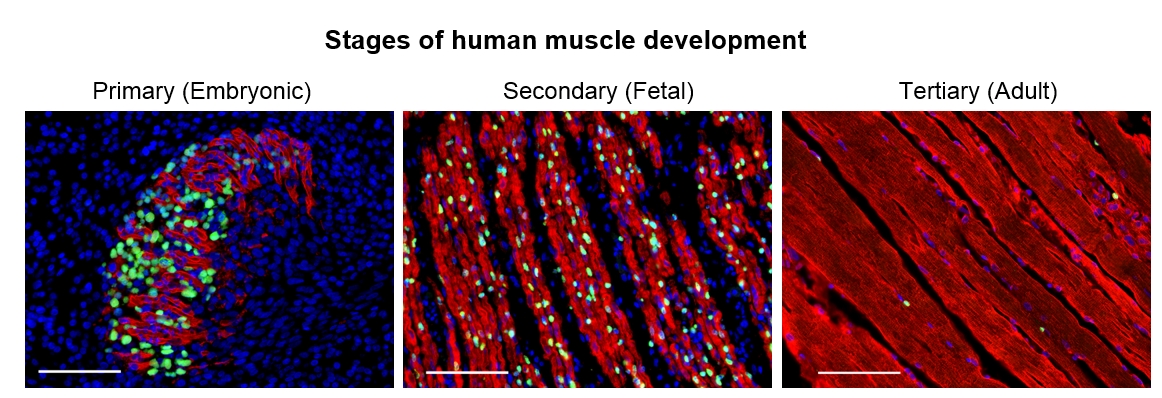
We set out to determine the myogenic potential of muscle cells created from hPSCs (termed skeletal muscle progenitor cells or hPSC-SMPCs), compared to the muscle cells from human embryonic, fetal, and adult myogenesis (Figure 1). Cells were dissociated from human tissues and their ability to fuse to form myotubes were compared to hPSC-SMPCs. We found that hPSC-SMPCs made from two commonly used directed differentiation protocols created muscle in a dish that looked and fused similarly to the embryonic muscle from primary myogenesis.
When we transplanted hPSC-SMPCs into mice, we found they did not engraft very well. Alternatively, human fetal cells from secondary myogenesis were very capable of forming new muscle in vivo.
Directed differentiation to skeletal muscle produces a number of heterogeneous cell types, and we thought that enrichment for the best-known adult satellite cell marker, known as NCAM or CD56, would improve our ability to engraft cells. However, NCAM enrichment failed, and if anything was slightly worse than unsorted hPSC-SMPCs. This was an example of the failure of applying adult tissue paradigms to immature hPSC progeny from directed differentiation. It was like comparing apples and oranges. After many months, we were no closer to making an engraftable cell from hPSCs.
Development guides the enrichment of functional muscle progenitors from hPSCs
We thought about how muscle cells created from hPSCs functionally resembled the morphology of muscle from embryonic myogenesis, but how muscle cells from fetal myogenesis were able to engraft very well. We hypothesized rather than trying to immediately create an adult cell from hPSC-SMPCs, we still had to direct their differentiation through more stages of human myogenesis and maturation.
The first thing we needed to do was determine the maturational identity of our cells. We performed RNA sequencing of NCAM+ hPSC-SMPCs, and compared gene expression to NCAM+ fetal week 17 SMPCs. As predicted, we found that hPSC-SMPCs were more embryonic than fetal SMPCs, but they were also more differentiated with increased expression of genes such as myogenin. This genetic profile supported the idea that the cells we were making more closely resembled the primary myogenic wave in which founder cells differentiate to establish the first muscle fibers, before a second wave of muscle progenitors arise that go on to make the adult satellite cells. Because NCAM is expressed by many cells in directed differentiation, including immature mesoderm cells, neurons, and more differentiated myoblasts, we hypothesized this was also a reason for the lack of improved enrichment and engraftment in vivo by NCAM+ hPSC-SMPCs.
We turned to our colleagues, Drs. Denis Evseenko and Ben Van Handel at the University of Southern California, who were facing a similar challenge with directed differentiation of hPSCs to chondrocytes. To identify unique genes of human chondrocytes, Dennis and Ben had performed a Weighed Gene Analyses (WGCNA) on five types of human fetal tissues including bone, cartilage, tendon, ligament, and skeletal muscle. In the fetal muscle dataset, they identified over 400 cell surface genes and transcription factors unique to skeletal muscle (Furgeson et al., 2018). A fun part of this project was pilfering through the gene data, making candidate lists, performing PubMed searches and refining cell surface genes to about 20 candidates. We then set up directed differentiations to evaluate new and known cell surface marker expression from the candidates in hPSC-SMPCs. From this screen, we found enrichment for ERBB3 or NGFR produced the most myogenic cell in vitro with robust fusion ability.
Eureka! ERBB3 or NGFR demarcate myogenic cells in human development
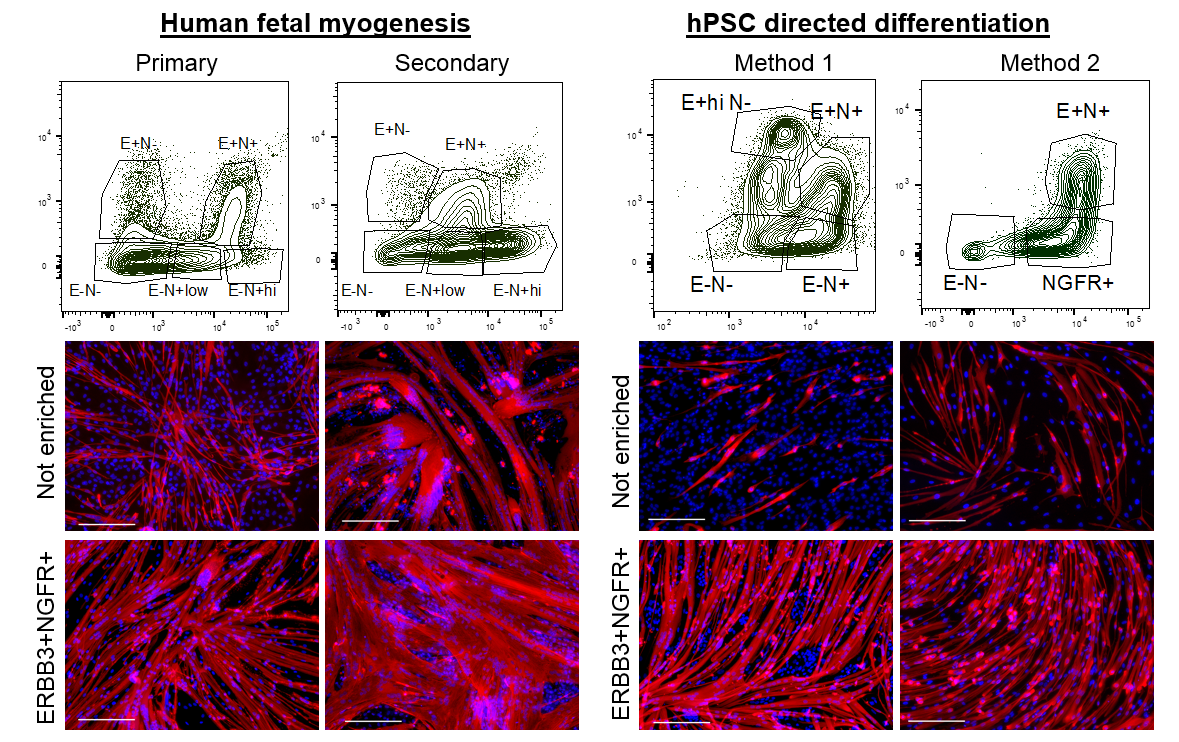
We wondered about the relevance of ERBB3 or NGFR in human development and whether these two cell surface markers demarcated the same cell or different cells at stages of development. We decided to analyze and enrich for cells based on ERBB3 or NGFR co-expression in primary and secondary myogenesis. We identified multiple subpopulations based on the expression profiles of these two cell surface receptors. In all cases, double positive ERBB3+NGFR+ cells resulted the highest PAX7 gene expression. Furthermore, primary and secondary myogenesis could be distinguished based on these markers, where very high expression marked a more immature progenitor (Figure 2). ERBB3 was not expressed by adult satellite cells.
We next applied this same enrichment strategy to hPSC-SMPCs. As expected, in every directed differentiation protocol tested double positive ERBB3+NGFR+ SMPCs could be induced to form homogeneous myotubes. These data demonstrate a striking similarity between the surface marker phenotypes of fetal myogenic progenitors and hPSC-SMPCs undergoing directed differentiation.
Development guides the maturation of muscle progenitors from hPSCs
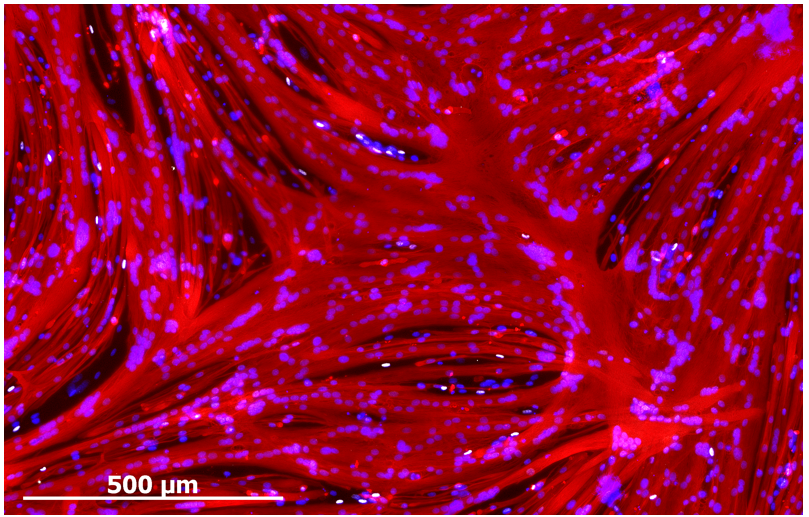
Despite enabling myogenic enrichment, differentiation of ERBB3+NGFR+ populations isolated from hPSCs yielded myotubes that were thinner and contained fewer nuclei than later stage fetal or adult counterparts. To understand the basis for these differences, we further analyzed our RNA sequencing data comparing hPSC-myotubes and fetal myotubes to their progenitors. These data revealed that activators of TGF-β signaling decreased during the course of fetal myotube differentiation. In contrast, hPSC-SMPCs had higher expression of TGF-β signaling genes and hPSC myotubes failed to down regulate TGF-β signaling, such as myostatin. When we blocked the TGF-β signaling we significantly increased hPSC-myotube fusion and produced morphology similar to late stage fetal myotubes (Figure 3). We performed multiple assays to verify that TGF-β inhibition indeed matured hPSC-myotubes.
ERBB3+ hPSC-SMPCs can generate new muscle fibers upon transplantation in vivo.
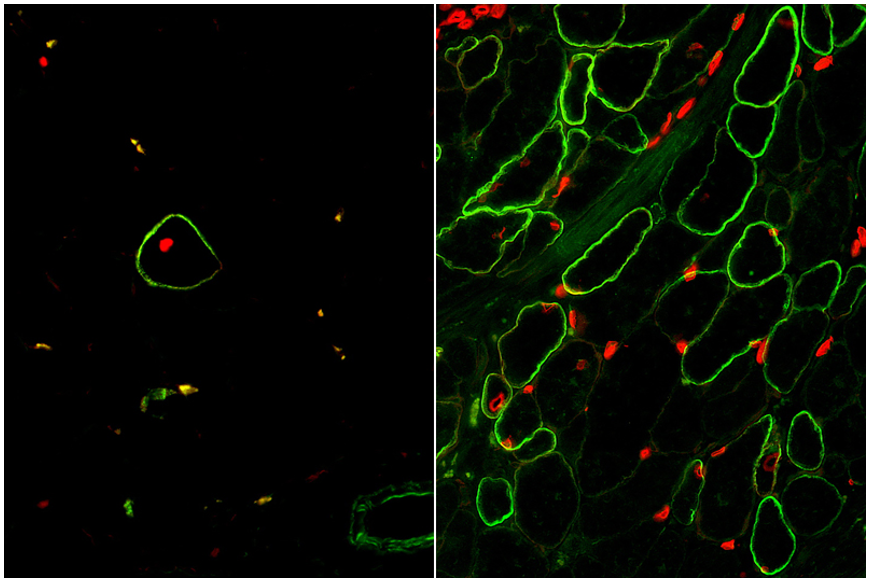
The moment of truth had arrived when our enrichment and maturation strategy was applied to hPSC-SMPC engraftment in vivo. After 30 days of waiting, mice injected with ERBB3+ hiPSC-SMPCs and treated with TGF-βi were evaluated for restoration of human dystrophin in a mouse model of DMD. We were gratified to find a greater than 50-fold improvement in number of dystrophin+ fibers using our directed differentiation strategy (Figure 4). This was especially satisfying because these cells were originally taken from boys with Duchenne muscular dystrophy, reprogrammed to induced pluripotent cells, and genetically modified with CRISPR/Cas9 to permanently correct the DMD gene mutation. What’s more, ERBB3+ hiPSC-SMPCs enabled engraftment approaching levels equivalent to directly-isolated human fetal muscle cells. By following human development, our results demonstrated a major advance in the ability to obtain engraftable SMPCs from hPSCs.
The next phase of this research will be to test and develop strategies to mature SMPCs to satellite cells similar to adult, which can reside in the stem cell niche in vivo and continuously repopulate new muscle after damage. We hope to continue to create a more regenerative cell for use in cell replacement therapy for patients with DMD and many other muscle wasting diseases.
References
Chal, J., et al., Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat Biotechnol, 2015. 33(9): p. 962-9.
Furgeson G et al., Transcriptional heterogeneity during lineage specification defines human skeletogenesis. In Review.
Hicks, M.R., et al., ERBB3 and NGFR mark a distinct skeletal muscle progenitor cell in human development and hPSCs. Nat Cell Biol, 2018. 20(1): p. 46-57.
Shelton M., et al., Derivation and expansion of PAX7-positive muscle progenitors from human and mouse embryonic stem cells. Stem Cell Reports, 2014. 3(3): p. 516-29.
Young C.S., et al., A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin functionin hiPSC-derived muscle cells. Cell Stem Cell, 2016. 18: p. 1-8.
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.