BSDB Gurdon Summer Studentship Report (4)
Posted by BSDB, on 20 November 2015
![]() In 2014, the British Society of Developmental Biology (BSDB) has initiated the Gurdon Summer Studentship program with the intention to provide highly motivated students with exceptional qualities and a strong interest in Developmental Biology an opportunity to engage in practical research. Each year, 10 successful applicants spend 8 weeks in the research laboratories of their choices, and the feedback we receive is outstanding. Please, read the student reports, kindly sent to us by Oliver Davis from Brighton and Sussex Medical School who was hosted in summer 2015 by Jean-Paul Vincent at the Crick Institute.
In 2014, the British Society of Developmental Biology (BSDB) has initiated the Gurdon Summer Studentship program with the intention to provide highly motivated students with exceptional qualities and a strong interest in Developmental Biology an opportunity to engage in practical research. Each year, 10 successful applicants spend 8 weeks in the research laboratories of their choices, and the feedback we receive is outstanding. Please, read the student reports, kindly sent to us by Oliver Davis from Brighton and Sussex Medical School who was hosted in summer 2015 by Jean-Paul Vincent at the Crick Institute.
Dying for a pattern
 This summer, I had the glorious opportunity of undertaking a BSDB funded research project in the laboratory of Jean-Paul Vincent at the Francis Crick Institute in Mill Hill, London. My project took place in the lab of Dr Jean-Paul Vincent (www.jpvincentlab.com) under the patient and inspiring tutelage of one of his PhD students, Sam Crossman. Our project investigated the mechanism of apoptosis in the model organism Drosophila melanogaster.Apoptosis is a form of programmed cell death and is an important process in the development of all multi-cellular organisms. Understanding why cells die in certain situations and not in others is of relevance to many areas of health, including embryological disorders and cancer, and my overall research aim was to investigate the role of apoptosis in the developing fly embryo. To do this, I worked with strains carrying mutations in genes required for patterning the anterior-posterior axis. Mutation of these so-called patterning genes can trigger extensive apoptosis in the embryonic epidermis (figure. 1) and therefore provides a useful model to investigate the apoptotic machinery in Drosophila.
This summer, I had the glorious opportunity of undertaking a BSDB funded research project in the laboratory of Jean-Paul Vincent at the Francis Crick Institute in Mill Hill, London. My project took place in the lab of Dr Jean-Paul Vincent (www.jpvincentlab.com) under the patient and inspiring tutelage of one of his PhD students, Sam Crossman. Our project investigated the mechanism of apoptosis in the model organism Drosophila melanogaster.Apoptosis is a form of programmed cell death and is an important process in the development of all multi-cellular organisms. Understanding why cells die in certain situations and not in others is of relevance to many areas of health, including embryological disorders and cancer, and my overall research aim was to investigate the role of apoptosis in the developing fly embryo. To do this, I worked with strains carrying mutations in genes required for patterning the anterior-posterior axis. Mutation of these so-called patterning genes can trigger extensive apoptosis in the embryonic epidermis (figure. 1) and therefore provides a useful model to investigate the apoptotic machinery in Drosophila.
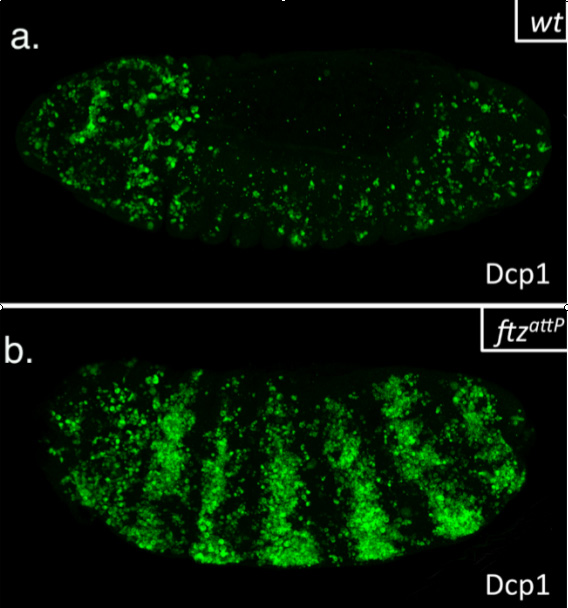
The cause of the ectopic apoptosis observed in patterning mutant embryos is not fully understood. One previously suggested explanation is that the cells of the epidermis can sense their ability to adopt the correct fate and undergo apoptosis if they lack the required patterning inputs to do so (Werz et al, 2005). However, if this were the case, there would have to exist an unknown machinery that would allow individuals cells to detect patterning errors and initiate apoptosis as a result.
In order to determine if cells are truly capable of detecting patterning errors, I planned to use a light inducible form of Cre recombinase to clonally remove a lox flanked allele of the ftz gene in a small subset of cells within each segment. If these small clones survive in an otherwise wild type embryo, it would argue against a cell-autonomous system where individual cells monitor their ability to differentiate correctly and would suggest that an alternative mechanism could be in play.
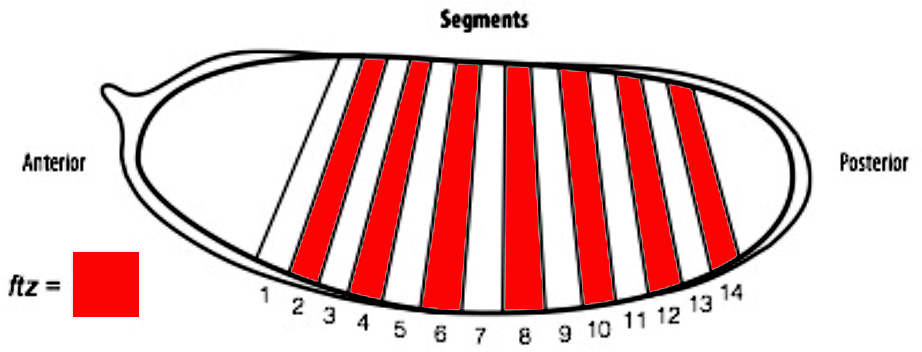
One difficulty with this plan is that ftz is activated very early on in embryogenesis. As a result, I set out to generate an early acting form of Cre, which could be used to remove ftz before it has carried out its function. To achieve this I spent the first part of my project cloning the Cre enzyme into a plasmid containing the actin promoter. As actin is an important protein in every cell, it is expressed from very early stages of embryogenesis. As a result, we hoped that by using the actin promoter to drive expression of Cre, we could produce the enzyme early enough to remove our lox flanked ftz allele in a timely manner and create mutant cells.
Unfortunately, cloning proved frustrating. Fortunately, it also proved educational. I learnt a lot about the way that experiments work, and how progress in science is more staccato than smooth. One issue I faced was the purification of my final plasmid using an Invitrogen maxiprep column. Each time I purified the plasmid I ended up with a lower yield than required, as I needed enough DNA to send to a company that would use it to generate a transgenic fly. I adjusted a parameter each time, but in the end it may have just been a plasmid with a low copy number as during my last attempt I purified it from a much larger bacterial culture, which finally gave me a sufficient amount of DNA.
The second part of my project was spent optimising a fluorescent in-situ hybridisation (FISH) protocol. I planned to use FISH to label cells expressing the pro-apoptotic gene hid in a series of patterning mutant embryos to characterise the regions where cell death occurs. As a control, I first conducted the protocol with a probe against the segment polarity gene wingless, which is expressed in a row of cells in every segment. This probe was made by someone else in the lab and is known to work, so I used it to learn the steps of the in-situ protocol.
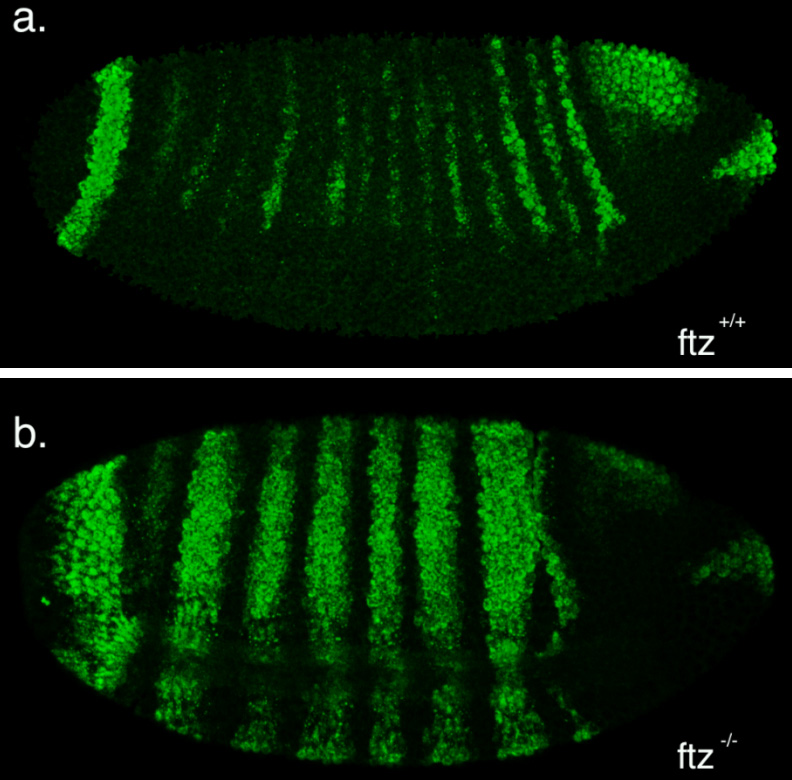
My FISH experiments with the wingless probe worked like a treat (figure 3), but difficulties soon followed when I attempted to make a new probe to label cells expressing hid. The stainings I conducted with the hid probe I had made repeatedly failed to work, and every attempt to generate a new probe proved unsuccessful. Disappointingly, I reached the end of my project before I managed to develop a protocol that worked, but I at least learnt plenty of science along the way!
My time in J.P.’s lab has been great for a number of reasons. I’ve come to love the problem solving nature of science and the freedom you get to explore what really interests you. It really is an adventure! However, I’ve also realised how difficult a career in science can be. Whilst this project has inspired me to become a scientist, I wonder if there’s a route into academia that would better suit my background as a medical student and my wider interests in medicine.
Sources
(1) Werz C, Lee TV, Lee PL, Lackey M, Bolduc C, Stein DS, Bergman A. Mis-specified cells die by an active gene-directed process, and inhibition of this death results in cell fate transformation in Drosophila. Development, 2005. 132(24): p. 5343-5352.
 (3 votes)
(3 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.