The Curious Case of Protocadherin 19 Epilepsy
Posted by Thomas Lab, on 12 March 2018
Daniel Pederick & Paul Thomas
Comment on our paper: Pederick, et al. 2018. Abnormal Cell Sorting Underlies the Unique X-Linked Inheritance of PCDH19 Epilepsy. Neuron 97 (1).
Here we discuss the curious case of female-restricted epilepsy, an unusual disorder caused by mutations in the Protocadherin 19 (PCDH19) gene. How changes in this cell adhesion gene cause seizures and intellectual disability in girls (but not boys) has been a mystery since this condition was first described over 20 years ago. By pursuing several lines of enquiry including in vitro cell sorting assays, CRISPR/Cas9 mouse models and patient MRIs, we have finally “cracked the case”, although intriguing questions remain about the neuronal pathology that underpins this unique condition.
PCDH19-GCE-no other disorder is quite like it
The most striking characteristic of PCDH19-GCE is its unique X-linked inheritance pattern which was first described by Ryan et al in 1997. Typically, X-linked disease-causing mutations affect males as they do not have a second wild type (WT) copy of the gene to compensate. As the name suggests, Protocadherin 19 Girls Clustering Epilepsy (PCDH19-GCE) is caused by mutation in the X-linked gene PCDH19 but this disorder does not follow the “typical” X-linked recessive disease inheritance pattern. Instead, heterozygous females (who have one WT copy and one mutated copy of the PCDH19) are affected while hemizygous males are not. A mysterious condition indeed!
It was not until 2008 that PCDH19 mutations were identified as causing the disease, and multiple hypotheses were put forward to explain its unusual inheritance pattern. It was initially proposed that PCDH19 mutations may be dominant negative, but the identification of whole gene deletions and examples of nonsense mediated decay provided strong evidence opposing this hypothesis (Dibbens et al., 2008). It was also suggested that males may be able to compensate for the loss of PCDH19 through the Y-linked PCDH11Y gene. However, this has also been ruled out due to the discovery of several mosaic males with early somatic mutations in PCDH19 that phenocopy affected girls (Depienne et al., 2009; Terracciano et al., 2016). The third and final hypothesis was based on two key facts. The first is that PCDH19 is located on the X-chromosome and is subject to X-inactivation. This mechanism ensures that females randomly “silence” one copy of the X chromosome in every cell to match the expression levels in males (who are XY). Thus, females with PCDH19 mutations have a mixture of WT and mutant neurons. The second was PCDH19’s known role as a homotypic cell adhesion molecule. Together these pieces of information led Dibbens et al , 2008 and Depienne et al 2009 to suggest that mosaic expression of PCDH19 in female brains leads to abnormal neuronal connections between PCDH19 WT and PCDH19 mutant cells affecting neural network formation and ultimately giving rise to seizures and intellectual disability. However, it was unclear how mosaicism could lead to PCDH19-GCE at the molecular, cellular and network level. Furthermore, this model did not explain why the complete absence of PCDH19 (in males) does not cause epilepsy.
The smoking gun: cell sorting assays provide evidence for a PCDH adhesion code
We attempted to unravel this mystery by combining complementary in vitro and in vivo experimental approaches. A valuable clue came from recent papers showing that clustered PCDHs (which are structurally similar to the non-clustered PCDH family to which PCDH19 belongs) act in combination to form complexes at the cell surface with highly specific homotypic binding affinities (Thu et al., 2014). This peaked our interest as it was unclear from the “cellular interference model” how the loss of a single cell adhesion molecule could disrupt normal interactions given that neurons typically express many PCDH family members. Using a co-immunoprecipitation and cell aggregation assays we demonstrated that PCDH19 can form promiscuous cis and highly specific trans interactions both of which are required to generate specific combinatorial binding affinities. We then mixed together two populations of cells expressing different combinations of PCDHs and observed segregation between the two populations when they differed by just a single PCDH (Figure 1). This confirmed that the “PCDH adhesion code” was acutely sensitive to the absence of PCDH19.
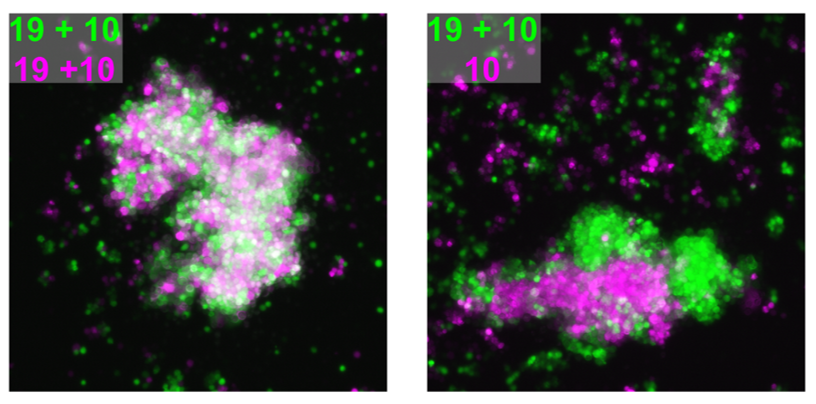
The next step was to assess how disruption of PCDH19-dependent adhesion would impact the developing brain, where a complex array of cell adhesion molecules and binding partners are expressed. To investigate this, we turned to our previously characterised Pcdh19 Knockout (KO) mouse model (Pederick et al., 2016). Like humans, Pcdh19 is located on the X chromosome in mice and therefore heterozygous mice have mosaic expression of Pcdh19 due to X-inactivation. Interestingly, electrocorticogram analysis revealed significantly elevated activity in heterozygous mice when compared to WT controls. Importantly, this phenotype was not present in mice completely lacking Pcdh19, which matched the unique X-linked inheritance of PCDH19 epilepsy.
CRISPR/Cas9 joins the investigation
While the electrical phenotype of the mouse was consistent with humans, we couldn’t investigate the cellular impact of mosaic PCDH19 expression in the brain because we were lacking a specific PCDH19 antibody with which to identify PCDH19+ cells. We therefore turned to the CRISPR/Cas9 genome editing system and generated a mouse with an epitope-tagged (HA-FLAG) version of PCDH19, allowing us to identify PCDH19 WT cells with commercially available antibodies. To detect “PCDH19-expressing” Pcdh19 null cells we used the previously mentioned PCDH19 KO mouse which has a LacZ reporter. Now we were set to answer the key question- how does mosaic expression of Pcdh19 in heterozygous female brains affect the behaviour of the WT PCDH19 and null PCDH19 cell populations?
Simultaneous labelling of WT PCDH19 and null PCDH19 cells in PCDH19 heterozygous mice revealed a striking pattern of alternating PCDH19 +ve and PCDH19 -ve regions (Figure 2). This pattern was particularly obvious in the developing cortex where it resembled “tiger stripes”. Before getting too excited, we needed to address the possibility that the apparent segregation simply arose from X-inactivation and subsequent clonal expansion of PCDH19 +ve and PCDH19 -ve cells. We therefore generated “WT” mice with one tagged copy of PCDH19 and one (untagged) WT PCDH19 allele. This critical control was only possible because we had generated the tagged allele (i.e. even if we had a PCDH19 Ab we could not have done this experiment). Critically, there was no sign of the “tiger stripes” in this control- instead we detected small patches of tagged and untagged PCDH19 cells along the ventricle with subtle variations in the cortical plate (Figure 2). So, evidence for active cell segregation in heterozygous females was beginning to stack up. Interestingly, the abnormal segregation pattern of WT PCDH19 and null PCDH19 cells was different in each mouse, presumably caused by the random nature of X-inactivation. Since the symptoms of PCDH19 epilepsy are highly variable (even in identical twins (Higurashi et al., 2012)) it is possible that different segregation patterns contribute to the severity of the disorder. Although not performed in this study it would be interesting to correlate the X-inactivation patterns seen in individual mice with their ECoG recordings to identify any patterns that lead to higher electrical brain activity.
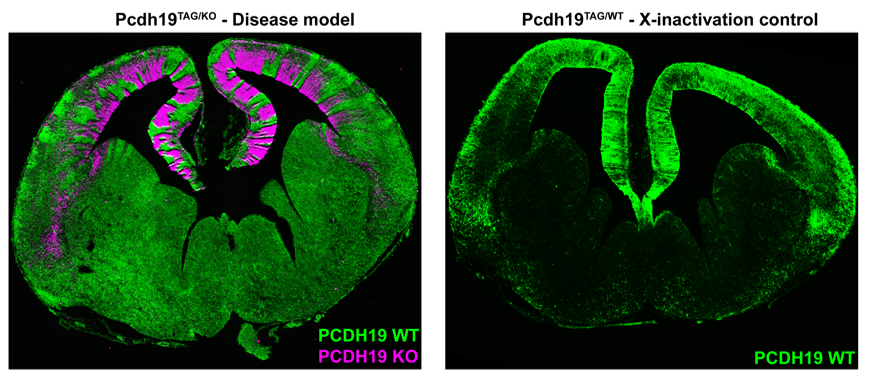
In our opinion the most interesting feature of PCDH19 epilepsy is not that mosaicism leads to disease but that individuals who completely lack PCDH19 do not have the disease. As mentioned above, homozygous null mice did not display increased electrical brain activity. We hypothesised that this phenotypic “rescue” was due to the uniform loss of Pchd19 and the consequent restoration of normal cell sorting during cortical development. To test this, we developed a strategy to rapidly assess in vivo cell sorting in Pcdh19 null embryos. We deleted the functional PCDH19TAG allele in heterozygous (PCDH19TAG/PCDH19LACZ) zygotes using CRISPR/Cas9, transferred the embryos to pseudopregnant females for further development, and then used X-Gal staining to track the location of the PCDH19 null cells. While negative (i.e. heterozygous) controls showed the expected segregation phenotype, no significant segregation occurred after deletion of the (WT) PCDH19TAG allele. These findings provided further evidence that abnormal cell sorting is caused by the differential adhesion affinities between WT PCDH19 and null PCDH19 cells. Furthermore, the absence of abnormal cell sorting in animals completely lacking PCDH19 provides a clear cellular phenotype that explains the unique inheritance pattern of PCDH19 epilepsy.
Finally, we postulated that abnormal cell sorting in humans could lead to brain malformations due to the prolonged expansion and increased cortical folding compared to mice. Abnormal cortical sulcation was observed in four girls with causative PCDH19 mutations. We identified variably positioned cortical defects that included bottom of the sulcus dysplasias, abnormal cortical folding, cortical thickening and blurring of the grey/white junction. It is not known how abnormal cell sorting may generate these cortical malformations, however, insight into the cellular mechanism could be gained by the generating a PCDH19 mutant ferret with CRISPR/Cas9 genome editing, a model organism that has extensive cortical folding.
For the future…
These findings provide some long-awaited answers to the mysterious inheritance of PCDH19 epilepsy. However, despite these insights, there is still much to discover about how mosaic expression of PCDH19 leads to epilepsy, intellectual disability and autism. It is possible that in addition to cell sorting, other processes such as neuronal wiring, synapse formation, maintenance and function are disrupted by differential adhesion affinities caused by mosaic expression of PCDH19. Perturbation of these fundamental neuronal processes is implicated in many neurodevelopmental disorders and it seems possible that they may also be altered in PCDH19 epilepsy. Much sleuthing lies ahead!
References
Depienne, C., Bouteiller, D., Keren, B., Cheuret, E., Poirier, K., Trouillard, O., Benyahia, B., Quelin, C., Carpentier, W., Julia, S., et al. (2009). Sporadic Infantile Epileptic Encephalopathy Caused by Mutations in PCDH19 Resembles Dravet Syndrome but Mainly Affects Females. PLoS Genet 5, e1000381.
Dibbens, L.M., Tarpey, P.S., Hynes, K., Bayly, M.A., Scheffer, I.E., Smith, R., Bomar, J., Sutton, E., Vandeleur, L., Shoubridge, C., et al. (2008). X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat. Genet. 40, 776–781.
Higurashi, N., Shi, X., Yasumoto, S., Oguni, H., Sakauchi, M., Itomi, K., Miyamoto, A., Shiraishi, H., Kato, T., Makita, Y., et al. (2012). PCDH19 mutation in Japanese females with epilepsy. Epilepsy Res. 99, 28–37.
Pederick, D.T., Homan, C.C., Jaehne, E.J., Piltz, S.G., Haines, B.P., Baune, B.T., Jolly, L.A., Hughes, J.N., Gecz, J., and Thomas, P.Q. (2016). Pcdh19 Loss-of-Function Increases Neuronal Migration In Vitro but is Dispensable for Brain Development in Mice. Scientific Reports 6, srep26765.
Ryan SG, Chance PF, Zou CH, Spinner NB, Golden JA, Smietana S. (1997) Epilepsy and mental retardation limited to females: an X-linked dominant disorder with male sparing. Nat Genet. 17, 92-5
Terracciano, A., Trivisano, M., Cusmai, R., De Palma, L., Fusco, L., Compagnucci, C., Bertini, E., Vigevano, F., and Specchio, N. (2016). PCDH19-related epilepsy in two mosaic male patients. Epilepsia.
Thu, C.A., Chen, W.V., Rubinstein, R., Chevee, M., Wolcott, H.N., Felsovalyi, K.O., Tapia, J.C., Shapiro, L., Honig, B., and Maniatis, T. (2014). Single-cell identity generated by combinatorial homophilic interactions between α, β, and γ protocadherins. Cell 158, 1045–1059.
 (3 votes)
(3 votes)3 thoughts on “The Curious Case of Protocadherin 19 Epilepsy”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
What a marvelous story. I was prompted to read this by a question asked by a student in my Human Genetics class. I was rewarded by this magnificent experimental odyssey.
I have a daughter with PCDH19 epilepsy and intellectual disabilities and this was such an interesting read. Thank You!
Thank you, it’s very interesting. Cheers from Yekaterinburg, Russia, and merry Xmas!