FISHing fish
Posted by NLVastenhouw, on 15 February 2016
By L. Carine Stapel and Nadine L.Vastenhouw
In developmental biology, the ability to analyze gene expression patterns is essential to address questions about gene regulation and cell fate. In our lab, we use zebrafish embryos to ask questions such as how is the onset of transcription in an embryo temporally regulated? How do cells in an embryo coordinate their transcriptional output? And what role does chromatin architecture play in regulating transcription? To address these questions in a quantitative manner, we need to determine exactly how many transcripts of a certain gene are present in any given cell at any given time. However, until recently, tools were lacking to allow automated detection and quantification of single RNAs at cellular resolution in zebrafish.
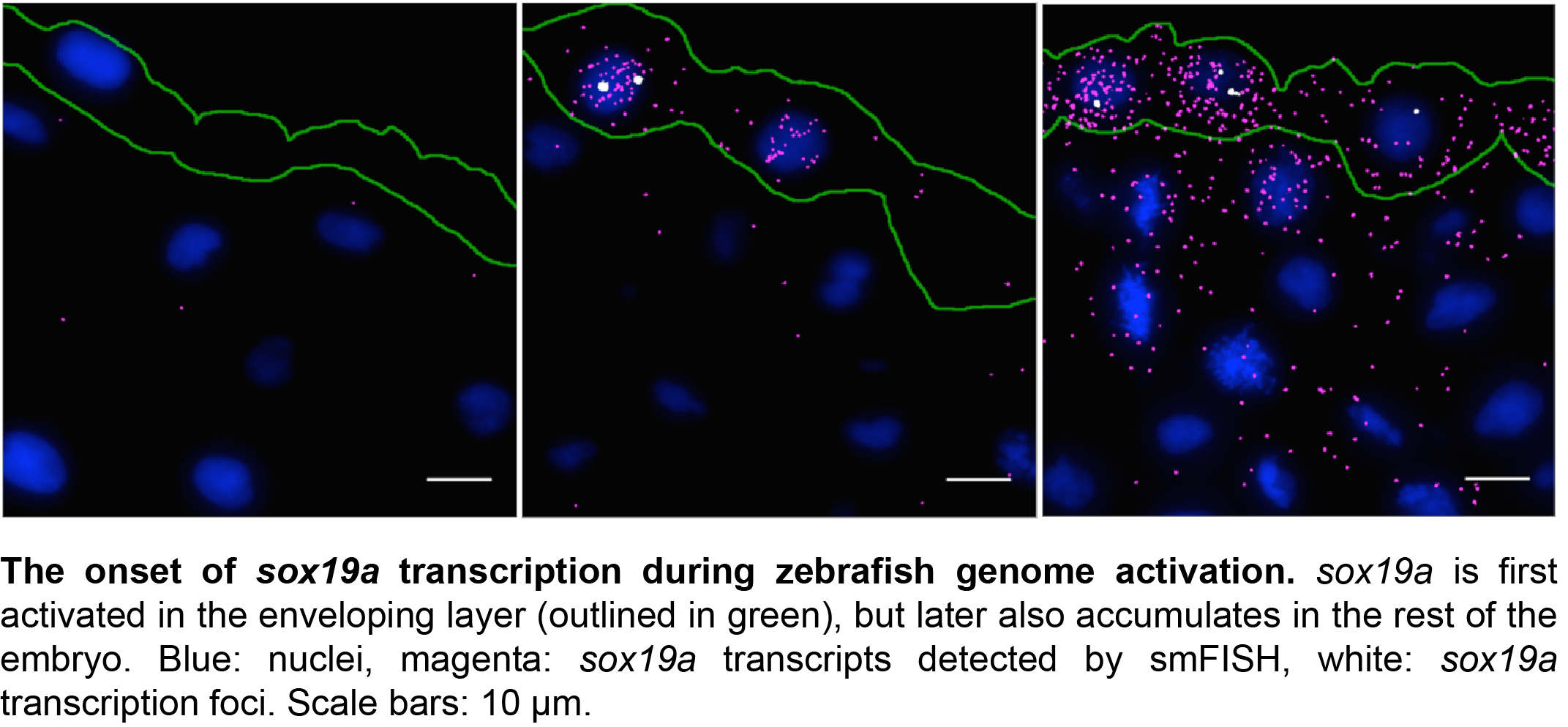
Spatial patterns of gene expression are typically studied by RNA in situ hybridization. However, the standard biotin- or digoxigenin-labeled RNA probes used in this technique are prone to diffuse localization, low sensitivity, and non-specific probe binding. Moreover, probe detection relies on antibody binding for visualization and is generally not quantitative1-3. Relative levels of gene expression can be studied using RNA sequencing approaches, but the technique has a very low sensitivity at the cellular level, as it only detects the ~10% most abundant transcripts4-6. Hope can nevertheless be found in the recently developed single molecule fluorescence in situ hybridization (smFISH) technique, which circumvents the above difficulties and detects transcripts at single-molecule resolution7,8.
The power of smFISH lies in the use of many short oligonucleotide probes that target a single mRNA transcript7,8. Each oligo is conjugated with one or more fluorophore. While the binding of multiple oligos to the same transcript yields a bright fluorescent spot, one oligo yields just a faint signal. Because mis-bound probes are unlikely to co-localize, this results in a high signal to noise ratio. Moreover, the short probes efficiently penetrate through target tissue, allowing for the detection of even low-abundant transcripts. Although smFISH has been used successfully both in single cells and tissues7,9-19, broad application of smFISH in complex samples has been hampered by the limited availability of protocols for embryos and by the lack of an automated image analysis pipeline that combines transcript detection with cell segmentation9,12,15,18. So we set out to develop a protocol for smFISH on embryo sections, and an analysis pipeline for automated transcript detection and cell segmentation20.
With support from Jan Philipp Junker and Alexander van Oudenaarden, first at MIT and later at the Hubrecht Institute in Utrecht, we expected the adaptation of an existing protocol to zebrafish embryos to be a gentle way into a PhD project. But as these things go, there were quite some hurdles that had to be overcome before we were able to produce reliable results. One major challenge was the production of good sections from early zebrafish embryos. This was overcome by serendipity. One week, when Carine was sick and unable to remove fixed embryos from their pre-embedding medium, we discovered that a long bath in pre-embedding medium results in the best sections further down the line. With this step included in our protocol, we were able to obtain specific, high-resolution information on gene expression for multiple genes simultaneously in zebrafish embryos at various stages of development. The freely available software package we developed detects these transcripts, as well as high-intensity foci corresponding to sites of active transcription in the nucleus.

We then collaborated with the lab of Gene Myers here at MPI-CBG to develop a semi-automated segmentation pipeline based on phalloidin staining of the membrane. With this tool, it now takes us 5 minutes per image to correct errors in the automated segmentation, in contrast to the 20 minutes a complete manual segmentation would take. While this is a major time saver, we are currently looking into the implementation of assisted manual correction, which will further reduce segmentation times. Using the automated pipeline, we can now calculate transcript densities in cells, nuclei and transcription foci for many cells with a simple mouse click.
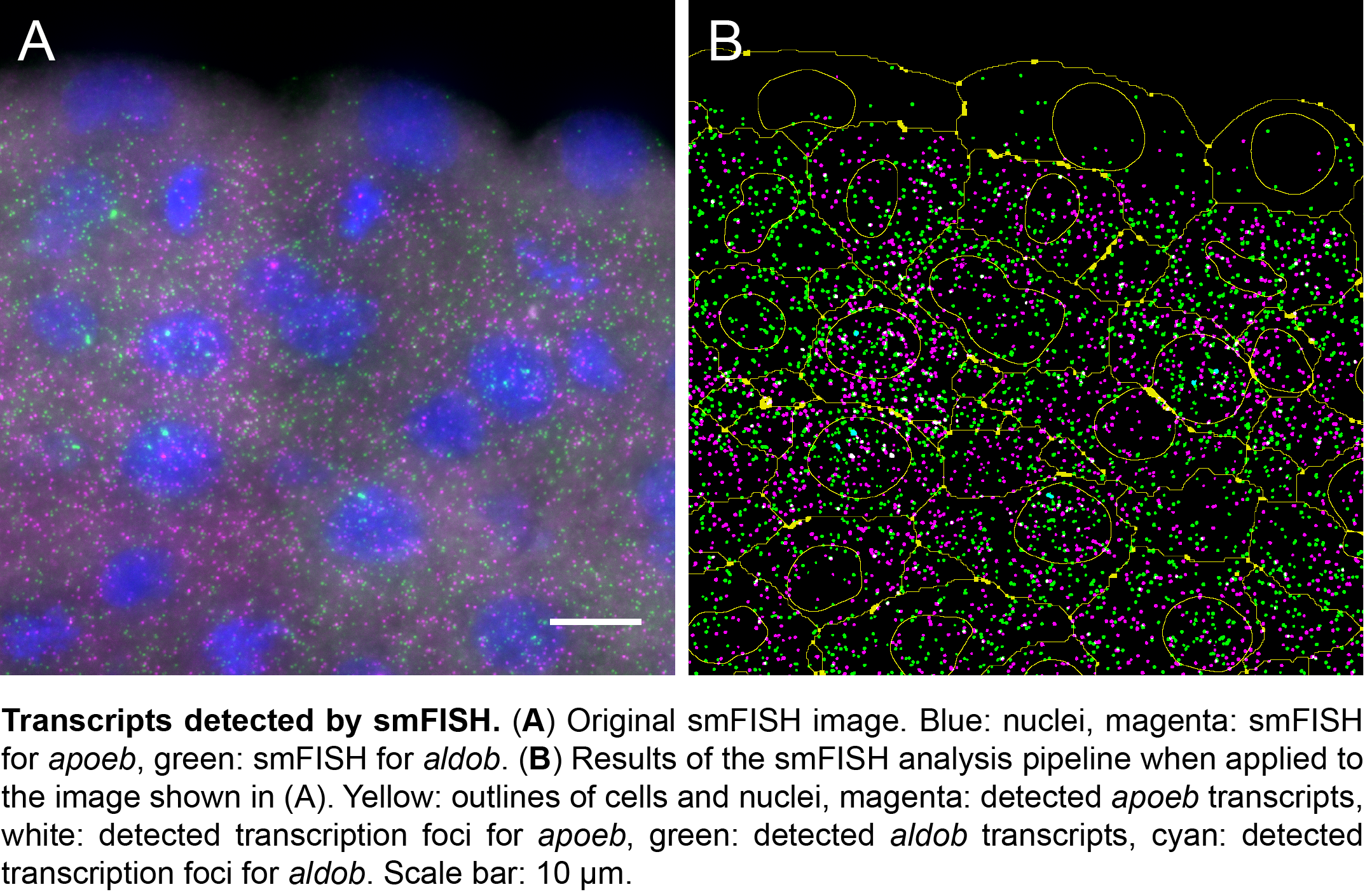
Considerable progress in sequencing technologies makes it possible to study the transcriptomic landscape of single cells. To open up these technologies for questions in developmental biology, efforts like TOMO-Seq5 and Seurat6 combine high-throughput transcriptome measurements in embryos with ways to preserve important spatial information about tissue context. Neither of these techniques, however, provides cellular resolution. Moreover, single cell sequencing is not (yet) sensitive enough to detect all transcripts in a cell, limiting its use to the most highly expressed genes. Thus, the smFISH protocol and image analysis pipeline that we published in Development completes the toolkit of biologists interested in spatially resolved gene expression at high resolution. Our custom-made software is freely available in KNIME (http://tinyurl.com/KNIME-MS-ECS) and Fiji (http://fiji.sc/MS-ECS-2D), so researchers working with a range of complex tissues can now start to exploit the benefits of high-resolution transcript analysis.
REFERENCES
- Thisse, C. & Thisse, B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3, 59–69 (2008).
- Tomancak, P. et al. Global analysis of patterns of gene expression during Drosophila embryogenesis. Genome Biol 8, R145 (2007).
- Gross-Thebing, T., Paksa, A. & Raz, E. Simultaneous high-resolution detection of multiple transcripts combined with localization of proteins in whole-mount embryos. BMC Biology 2014 12:1 12, 1 (2014).
- Grün, D., Kester, L. & van Oudenaarden, A. Validation of noise models for single-cell transcriptomics. Nat Meth 11, 637–640 (2014).
- Junker, J. P. et al. Genome-wide RNA Tomography in the zebrafish embryo. Cell 159, 662–675 (2014).
- Satija, R., Farrell, J. A., Gennert, D., Schier, A. F. & Regev, A. Spatial reconstruction of single-cell gene expression data. Nat. Biotechnol. 33, 495–502 (2015).
- Raj, A., van den Bogaard, P., Rifkin, S. A., van Oudenaarden, A. & Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat Meth 5, 877–879 (2008).
- Femino, A. M., Fay, F. S., Fogarty, K. & Singer, R. H. Visualization of single RNA transcripts in situ. Science 280, 585–590 (1998).
- Bahar Halpern, K. et al. Bursty gene expression in the intact Mammalian liver. Molecular Cell 58, 147–156 (2015).
- Battich, N., Stoeger, T. & Pelkmans, L. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nat Meth 10, 1127–1133 (2013).
- Boettiger, A. N. & Levine, M. Rapid transcription fosters coordinate snail expression in the Drosophila embryo. CellReports 3, 8–15 (2013).
- Itzkovitz, S. et al. Single-molecule transcript counting of stem-cell markers in the mouse intestine. Nat Cell Biol 14, 106–114 (2011).
- Itzkovitz, S., Blat, I. C., Jacks, T., Clevers, H. & van Oudenaarden, A. Optimality in the Development of Intestinal Crypts. Cell 148, 608–619 (2012).
- Little, S. C., Tikhonov, M. & Gregor, T. Precise Developmental Gene Expression Arises from Globally Stochastic Transcriptional Activity. Cell 154, 789–800 (2013).
- Lyubimova, A. et al. Single-molecule mRNA detection and counting in mammalian tissue. Nat Protoc 8, 1743–1758 (2013).
- Mueller, F. et al. FISH-quant: automatic counting of transcripts in 3D FISH images. Nat Meth 10, 277–278 (2013).
- Nair, G., Walton, T., Murray, J. I. & Raj, A. Gene transcription is coordinated with, but not dependent on, cell divisions during C. elegans embryonic fate specification. Development 140, 3385–3394 (2013).
- Oka, Y. & Sato, T. N. Whole-mount single molecule FISH method for zebrafish embryo. Sci. Rep. 5, 8571 (2015).
- Peterson, K. A. et al. Neural-specific Sox2 input and differential Gli-binding affinity provide context and positional information in Shh-directed neural patterning. Genes & Development 26, 2802–2816 (2012).
- Stapel, L. C. et al. Automated detection and quantification of single RNAs at cellular resolution in zebrafish embryos. Development 143, 540–546 (2016).
 (9 votes)
(9 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.