From fate to form: the journey to whole-embryo spatial transcriptomics
Posted by Yinan Wan, on 2 April 2026
Where molecules meet form
I have long been interested in understanding how development emerges at the intersection of molecular and spatial organization. On one hand, decades of work have identified the genes and signaling pathways that control cell fate. On the other hand, classical embryology and biophysics have revealed how cells move, change shape, and assemble into tissues. Yet, directly connecting these two layers—linking gene expression to spatial patterns and morphogenesis at the scale of a whole embryo—has remained challenging.
When I joined the Schier lab in March 2020, single-cell RNA-seq approaches had already enabled the reconstruction of developmental trajectories with remarkable molecular detail [1]. For the first time, we could computationally line up cells along developmental paths and begin to understand how cell fates emerge at the whole-embryo level. But something always felt missing to me: these trajectories existed in abstract space, detached from the physical embryo. We could describe where cells were going, but not where they actually were.
At the same time, there was growing interest in spatial transcriptomics within the lab and through our membership in the Allen Discovery Center for Cell Lineage Tracing. This created an opportunity to bridge molecular and spatial information in developing systems. With my background in imaging and technology development, I was particularly drawn to the idea of building a method that could map gene expression across whole embryos, while preserving spatial organization at high resolution.
Technically, the initial setup went relatively smoothly. Ahilya Sawh (then in Susan Mango’s lab at the Biozentrum, now leading her own group at the University of Toronto, Canada) had previously established a FISH-based system for chromosome tracing in C. elegans [2, 3]. With support from the Biozentrum Imaging Core Facility, we adapted this system onto a Nikon microscope, making it more accessible for biological applications. Having this foundation in place was reassuring—it meant that the challenge ahead was not starting from zero, but rather pushing something promising to its limits.

Making it work in a whole embryo
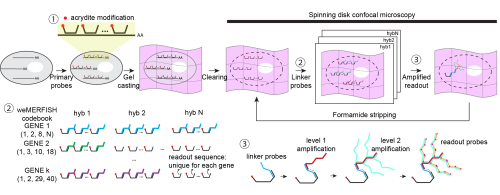
Early on, we realized that if we wanted to understand gene expression in a whole embryo, measuring just a handful of genes would not be enough. We needed to look at hundreds of genes at the same time, but without an impractical number of imaging rounds. This led us to explore MERFISH, which uses a combinatorial barcode system to identify genes across multiple rounds of imaging and allows many genes to be read out efficiently [4, 5]. In practice, it felt like a good balance between scale and feasibility for what we wanted to do.
While the foundation was in place, adapting MERFISH to whole embryos still required several key innovations. An important part of this effort was our close collaboration with Bogdan Bintu at UCSD in the USA. During his time in Xiaowei Zhuang’s lab at Harvard University, Bogdan developed high-throughput imaging approaches that combine MERFISH with 3D chromatin organization [6], contributing deep experience in both experimental and computational aspects. He had already begun implementing important technical improvements and generously shared his strategies with us, as well as providing essential support for the instrumentation and computational pipeline.
In the end, making this work required combining several key improvements, each addressing a different limitation of the system:
• Stability during long-term imaging
Whole-embryo MERFISH imaging can take weeks, as the sample must be imaged plane by plane, region by region, and across multiple hybridization rounds. Early on, this long-term imaging became one of the most frustrating bottlenecks. Using conventional approaches, in which mRNAs are anchored to a gel and repeatedly probed, we saw signals gradually fading over time. Even under carefully controlled RNase-free conditions, degradation was unavoidable.
The solution came from rethinking the problem: instead of anchoring RNA, we anchored the primary DNA probes using acrydite modifications. This seemingly simple shift made a huge difference. The signal remained stable even after a month of continuous imaging, and we could use harsh stripping conditions (80–100% formamide) without worrying about losing the probes. It was one of those moments where a technical fix suddenly makes everything feel possible.
• Seeing deeper without losing signal
Imaging deep tissues introduced another challenge. Spinning disk confocal microscopy is essential for thick samples, but it comes with reduced signal compared to widefield imaging. We initially explored computational approaches to recover signal, but in practice, signal and noise often looked too similar to confidently separate.
Instead, we turned to a physical solution: branching amplification. By boosting the signal at the molecular level, we were able to image more reliably, reduce exposure time, and make the data much more interpretable, especially in deeper regions of the embryo.
• Designing for flexibility
Another challenge was making the method usable in practice. Traditional MERFISH requires encoding the combinatorial barcode directly into the primary probes, effectively locking the experiment into a fixed readout strategy.
We wanted something more flexible. By introducing a gene-specific linker system, we decoupled probe design from readout. This means that researchers can design large probe libraries first, and decide later how to read them out—sequentially for a few genes, or combinatorially for many. This flexibility turned out to be important not just technically, but psychologically: it lowers the barrier to trying the method in the first place.
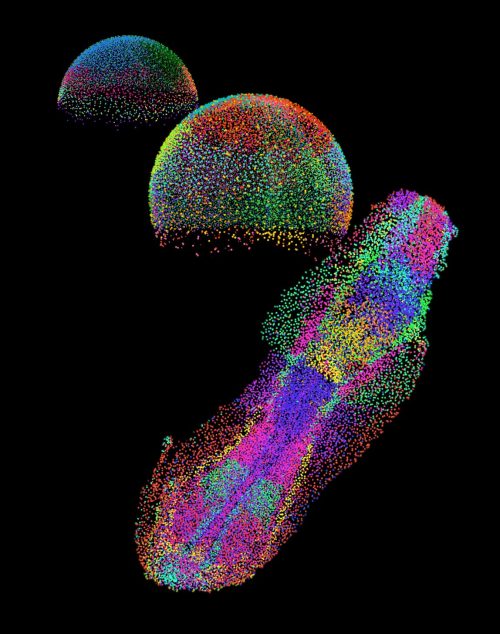
When the data started to speak
Once we had the data, the analysis moved quickly. I remember feeling a mix of excitement and disbelief: after spending three years building the system, suddenly everything was there at once. The work was tremendously accelerated by the incredible team behind it: Jakob El Kholtei and I co-developed the weMERFISH method and the data processing pipeline. KJ Jenie, an exceptionally talented undergraduate at the time in Bogdan’s lab, built the MERFISHEYES website (https://schier.merfisheyes.com), making the dataset accessible and explorable. A key component of the project came from my postdoctoral colleague Jialin Liu, who had generated a comprehensive scMultiomics dataset of zebrafish development to study the regulatory logic of cell type specification [7], and very generously made it available to us. Integrating his scMultiomics data with the weMERFISH data allowed us to comprehensively map gene expression and chromatin accessibility in space, creating a multiomic atlas. Mariona Colomer-Rosell performed the analysis of these multiomic data and helped uncover principles of tissue-specific gene regulation at the whole-embryo level. Bringing together these different pieces was essential for turning the dataset into something we could truly interpret.
One of the most fascinating aspects for me was the concept of “time” in development. In single-cell data, we often reconstruct “pseudotime” trajectories, but seeing these trajectories mapped into real space was incredibly satisfying. Along the zebrafish tail, for example, we could directly observe the progression from progenitors to differentiated cells as a spatial gradient. It was one of the first moments where the abstract and the physical truly aligned.
We also applied a spatial version of RNA velocity [8, 9], using nuclear versus cytoplasmic transcripts as a proxy for transcriptional dynamics. What surprised us was that, especially in early development, the inferred transcriptional dynamics mirrored physical cell movements during morphogenesis. At first glance, this feels intuitive. But the underlying mechanisms are very different: transcriptional regulation and cell movement are controlled by distinct processes. The fact that they align so closely suggests a deeper coupling between gene expression dynamics and morphogenesis. This was one of those observations that stayed with me, because it hints at something fundamental that we don’t yet fully understand.
Another memorable part of the journey was the path from preprint to publication. When we first posted the work on bioRxiv and launched the MERFISHEYES website, the response was immediate and very encouraging. People started exploring the data, reaching out with questions, and even visiting the lab to learn how to set up the method. Seeing the dataset being used so quickly made us realize that it could become a resource for the community much earlier than we had expected.
At the same time, the peer review process pushed the work in important ways. The reviewers appreciated the technology and the dataset, but also challenged us to go further, especially to better connect the method to biological questions and to take fuller advantage of the multimodal data. Addressing these comments led to substantial additions and improvements throughout the paper. We expanded the analysis of subcellular transcript localization, strengthened the RNA velocity framework, benchmarked data integration methods more rigorously, and added new analyses such as cell–cell communication.
Perhaps most importantly, the revision motivated us to develop MERFISH-FATE in collaboration with Guoqiang Yu’s group (Tsinghua University, China), integrating spatial transcriptomics with live imaging to directly link gene expression changes to morphogenetic movements. Specifically, we mapped corresponding regions between a weMERFISH embryo and a live-imaged embryo at early gastrulation, where cells had been tracked throughout development. We then followed these trajectories forward and mapped the descendant cells back to their corresponding regions at mid-gastrulation, effectively connecting gene expression patterns across time.
This became a great extension of the story and shifted the paper from a largely static atlas into a more dynamic view of development. We then spent months simply looking at how patterns evolve—scrolling through images, comparing stages, trying to build intuition. Across many genes, we saw surprisingly complex dynamics. One example is tbxta, which is expressed at the embryonic margin at both early and mid-gastrulation. It would be natural to assume this reflects simple inheritance. But when we incorporated cell dynamics, we found that some cells activate tbxta while others turn it off. What looked like a static pattern was actually the result of dynamic and opposing processes. Moments like this made me appreciate how much information is lost when we only look at snapshots, and how powerful it is to connect gene expression with cell behavior. This is a direction we are now continuing to explore in more depth in a recent preprint describing fate mapping in zebrafish embryogenesis and beyond [10].
Looking ahead: from description to understanding
weMERFISH provides a way to map gene expression and cellular states across intact, developing tissues in 3D. For me, a particularly exciting direction is to extend this approach to larger and more complex systems: whole organs, organoids, and beyond. Moving into these systems will allow us to study how spatial gene expression patterns scale with size and geometry, and how these patterns are adapted across evolution.
At the same time, a major direction in the field, in my view, is the integration of multiple modalities. In this work, we combined weMERFISH with chromatin accessibility and embryo morphogenesis, and this naturally raises broader questions: how does gene expression relate to 3D genome organization? To protein distribution? To lineage history and cell behavior? Each of these layers captures a different aspect of cellular identity, and I believe only by combining them can we begin to understand how cells make decisions in their native context.
This is also a direction I am eager to pursue in my own future work. These multimodal datasets are not just richer—they fundamentally change the type of questions we can ask. Instead of only describing patterns, we can begin to build models that explain how these patterns arise. We can start to ask causal questions: which molecular features predict a cell’s future behavior? Which spatial contexts bias fate decisions? And how conserved are these relationships across tissues and embryos?
Ultimately, I am particularly interested in understanding how variability between individual cells gives rise to robust and reproducible tissue structures. Development is remarkably reliable, despite underlying variability. Technologies like weMERFISH bring us closer to uncovering these principles, and to understanding how a single fertilized egg reliably gives rise to a complex organism.
Access the article:
Wan, Y., J. El Kholtei, I. Jenie, M. Colomer-Rosell, J. Liu, Q. Zhang, J. Navajas Acedo, L. Y. Du, M. Codina-Tobias, M. Wang, W. Zheng, E. Lin, T. H. Chuang, O. Mayseless, A. Sawh, S. E. Mango, G. Yu, B. Bintu, and A. F. Schier. 2026. “Whole-embryo spatial transcriptomics at subcellular resolution from gastrulation to organogenesis.” Science 391 (6790): eadt3439. https://doi.org/10.1126/science.adt3439.
References
- Farrell, J.A., et al., Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 2018. 360(6392).
- Sawh, A.N., et al., Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. elegans Embryos. Mol Cell, 2020. 78(1): p. 96–111 e6.
- Sawh, A.N. and S.E. Mango, Multiplexed Sequential DNA FISH in Caenorhabditis elegans Embryos. STAR Protoc, 2020. 1(3): p. 100107.
- Chen, K.H., et al., RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science, 2015. 348(6233): p. aaa6090.
- Moffitt, J.R. and X. Zhuang, RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol, 2016. 572: p. 1–49.
- Su, J.H., et al., Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell, 2020. 182(6): p. 1641–1659 e26.
- Liu, J., et al., Decoding the regulatory logic of specification and differentiation during vertebrate embryogenesis. bioRxiv, 2024.
- La Manno, G., et al., RNA velocity of single cells. Nature, 2018. 560(7719): p. 494–498.
- Bergen, V., et al., Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol, 2020. 38(12): p. 1408–1414.
- Wang, M., et al., High-Fidelity Long-term Whole-embryo Lineage and Fate Reconstruction by Iterative Tracking with Error Correction. bioRxiv, 2026: p. 2026.03. 12.711203.
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.