A glimpse of a dynamic cell fate decision
Posted by Andras, on 27 October 2017
Andras Paldi, Daniel Stockholm, Alice Moussy
How do phenotypic differences between cells of the same clonal origin emerge? How exactly does the transition between the initial and final phenotypes occur? What happens in the cell during the transition? When there are two or more options, how is the choice made between them? How long does it take to acquire a new phenotype? What is the minimal difference required to consider two cells as phenotypically different? To define a cell type, should we consider only morphological, molecular differences or both simultaneously?
Despite the plethora of studies, these simple questions remain unanswered. Most of the studies focus on identifying the essential genes or environmental factors usually at the level of cell populations. The huge amount of molecular data accumulated over the past decades gave us the illusion of knowledge. Knowing the players is obviously essential, but this is only the starting point for the understanding. Unfortunately, our understanding of the process of differentiation remains desperately scarce, as the lack of clear responses to the above listed simple questions shows.
Recently, two important challenges came to modify the perception of differentiation. The first comes from the spectacular development of single-cell technologies. The second is that we have to come to realize how much phenotypic plasticity is a genuine characteristic of cells. The main lesson from the rapid development of single-cell detection techniques is the unambiguous demonstration of how different individual cells are and how poorly population-level averages represent them. The bulk of our knowledge on differentiation comes from studies of cell populations. Perhaps unconsciously, we took for granted that individual cells all follow with small variation the same sequence of events as what we could see at the level of cell populations. The unexpectedly high variation of individual cell phenotypes in populations that were believed to be homogenous (because of the morphological similarity of the cells, their clonal origin, their expression of some markers etc.) draw the attention to the phenotypic plasticity of the cells and to the fact that fate decisions are “taken” by individual cells.
Clearly, a coherent, systemic level explicative frame is needed that can account for the coherent population-level behaviour emerging from highly variable individual cell phenotypes and behaviour.
Our recently published work [1] was motivated by the wish to contribute to this effort. Although we used the extensively studied hematopoietic stem cell model, we were surprised how much these general questions fit to the model. The definition of the hematopoietic stem cell is widely debated, no precise description of the earliest events of differentiation and only very scarce information on the morphological changes during the same period were available. The study of hematopoietic stem cells is made difficult by the lack of exact criteria to identify them. Nonetheless, there is a consensus that CD34+ cell fraction in the umbilical cord blood contains high number of these cells. We decided therefore to privilege an integrated view and work on the whole population of CD34+ cells. Individual cells were randomly sorted from the population at different fixed time-points and their gene expression profile was analysed by single-cell RT-PCR. The structure of the population and its components were identified on the basis of the collected single-cell gene expression data. Parallelly, we set up a time-lapse system that allowed the continuous monitoring of the cells and their progeny during the first 96hrs after stimulation. Adding the continuous observations of the morphological changes and cell division timing of individual cells to the single time-point single-cell molecular analysis sampling of the same population provided a glimpse of the true dynamic nature of cellular fate decision.
The CD34+ cell fraction is traditionally considered as heterogeneous. Indeed, before cytokine stimulation every cell displayed a unique gene expression profile. However, no groups could be identified on the basis of their statistical similarity, this population is not a mixture of a limited number of “cell types”. When cytokines were added to the culture, every cell responded in a unique way. Again, every cell displayed a unique gene expression pattern that was different of the previous seen at t=0 hours. It was characterized by the simultaneous expression of different lineage-specific genes. This state is known as a multi-lineage primed pattern [2, 3]. A second round of change occurred during the second 24 hours. Two days after the stimulation of the cells two distinct transcription patterns emerged. One pattern was typical for myelo-erythroid progenitors, while the second was reminiscent of multipotent cells. Until now, the results overall confirmed the previous studies; the main novelty was the apparent rapidity of transition from the initial to a multilineage primed gene expression pattern and, just 24 hours later, to two distinct profiles. However, these snapshots did not allow us to deduce on the dynamics of the changes.

The real surprise came from the analysis of the time-lapse records. Individual CD34+ cells were placed in microwells and imaged for a week at 1 image/min. The resulting time-lapse records allowed us to record cell cycle lengths and morphological changes of each individual cell within individual clones. After stimulation, the cells usually displayed a polarized shape with a strong protrusion on one side called uropod. The first unexpected observation was to see that the unusual length of the first cell cycle. The cells made more than 50 hours on average to divide. This means, that the first major transcriptional change occurred during the first cell cycle and the second around the end of the first or the very beginning of the second cell cycle. After the first division, we could see two different morphologies; one was strongly polarized with a uropod, the second is spherical. The daughter cells usually inherited the morphology of their mothers. Polarized cells gave two polarized and round cells two round daughters. However, we were surprised to observe that a significant proportion of the cells did not conserve a stable morphology; they switched from one morphology to another and back many times during the cell cycle. The majority of their daughter cells also conserved the fluctuating phenotype
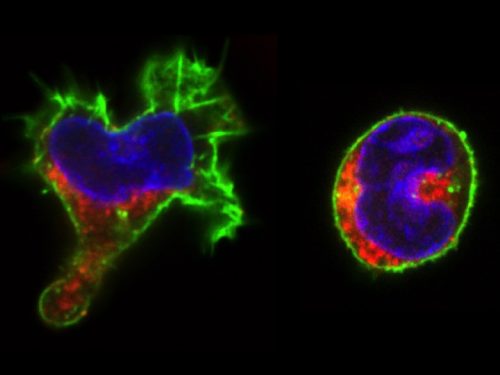
We called these cells “hesitant”. The overall picture suggest that stimulated CD34+ cells, after a brief passage through a multilineage primed state reach (without cell division!) one of the two alternative states characterized by a typical transcription pattern and cellular morphology. However, a significant proportion of cells fluctuate between the two morphologies. Does this morphological instability reflect transcriptome fluctuations? To answer this question, we have isolated individual cells with the three different – stable round, polarized or “hesitant” – behaviours and analysed their gene expression pattern. It appeared that the cells with stable morphology displayed one of the two expression patterns first observed at the 48 hors time point. The “hesitant” cells were characterized by an intermediate profile. The molecular analysis correlated to the time-lapse data suggests therefore that these cells are in an unstable state; their transcriptome undergoes fluctuations that are reflected in their fluctuating morphology also.
These are the key observations and the immediate conclusions of this work. However, these observations may contribute to the current tendency to reframe the issue of cell differentiation and stem cells in general. Cell differentiation can be approached using the concepts of stability and change – two complementary concepts widely used in biology. Stem cells may represent a highly unstable cell fraction contrary to the cells with stable differentiated phenotype. Unstable stem cells are actively exploring the space of available phenotypes before getting trapped by one of them. This is a kind of trial-and-error process. Under normal conditions, the unstable period is relatively short lasting; this is why the unstable cells we consider as stem cells are so rare in a normal tissue. However, a substantial change in the environment can destabilize many cells at the same time. This is what we see when CD34+ cells are stressed by the sudden addition of a cytokine cocktail. Due to the progressive adaptation to the new environment, the proportion of the “hesitant” stem cells decreases gradually as they attracted to the more adapted phenotypes. Importantly, this process seems to depend only indirectly on cell divisions. This interpretation is in remarkable agreement with earlier theoretical predictions and experimental work [4-8] and supported by recent experimental observations also [9-12].
Our paper was initially submitted to a well-known journal in stem cell biology. Beyond the disappointment of the rejections (every scientist is used to that), we were surprised by the poor quality of the reviews. The referees raised some technical concerns about the single-cell RT-PCR versus single-cell RNA sequencing, but not a single word about the time-lapse experiments that represented the major part of the paper, nor about the coupling the molecular and cellular scales, which is the true originality of the work. The reviews were very different when the manuscript was submitted to PloS Biology. The comments concerned all aspects of the work and the suggestions truly helped to improve the final version.
It would be naïve to think that a single paper can answer the fundamental questions raised at the beginning of this text. Clearly, we need a fresh view on cell differentiation that goes beyond the simple gathering and classification of molecular data and takes into account the true dynamics.
References
- Moussy A, Cosette J, Parmentier R, da Silva C, Corre G, Richard A, et al. Integrated time-lapse and single-cell transcription studies highlight the variable and dynamic nature of human hematopoietic cell fate commitment. PLoS Biol. 2017;15(7):e2001867. doi: 10.1371/journal.pbio.2001867. PubMed PMID: 28749943; PubMed Central PMCID: PMC5531424.
- Hu M, Krause D, Greaves M, Sharkis S, Dexter M, Heyworth C, et al. Multilineage gene expression precedes commitment in the hemopoietic system. Genes & development. 1997;11(6):774-85. PubMed PMID: 9087431.
- Pina C, Fugazza C, Tipping AJ, Brown J, Soneji S, Teles J, et al. Inferring rules of lineage commitment in haematopoiesis. Nature cell biology. 2012;14(3):287-94. doi: 10.1038/ncb2442. PubMed PMID: 22344032.
- Furusawa C, Kaneko K. A dynamical-systems view of stem cell biology. Science. 2012;338(6104):215-7. doi: 10.1126/science.1224311. PubMed PMID: 23066073.
- Huang S. Non-genetic heterogeneity of cells in development: more than just noise. Development. 2009;136(23):3853-62. doi: 10.1242/dev.035139. PubMed PMID: 19906852; PubMed Central PMCID: PMC2778736.
- Kupiec JJ. A chance-selection model for cell differentiation. Cell death and differentiation. 1996;3(4):385-90. PubMed PMID: 17180108.
- Kupiec JJ. A Darwinian theory for the origin of cellular differentiation. Molecular & general genetics : MGG. 1997;255(2):201-8. PubMed PMID: 9236778.
- Paldi A. Stochastic gene expression during cell differentiation: order from disorder? Cellular and molecular life sciences : CMLS. 2003;60(9):1775-8. doi: 10.1007/s00018-003-23147-z. PubMed PMID: 14523542.
- Mojtahedi M, Skupin A, Zhou J, Castano IG, Leong-Quong RY, Chang H, et al. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016;14(12):e2000640. doi: 10.1371/journal.pbio.2000640. PubMed PMID: 28027308; PubMed Central PMCID: PMC5189937.
- Notta F, Zandi S, Takayama N, Dobson S, Gan OI, Wilson G, et al. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351(6269):aab2116. doi: 10.1126/science.aab2116. PubMed PMID: 26541609; PubMed Central PMCID: PMC4816201.
- Richard A, Boullu L, Herbach U, Bonnafoux A, Morin V, Vallin E, et al. Single-cell-based analysis highlights a surge in cell-to-cell molecular variability preceding irreversible commitment in a differentiation process. . Plos Biology. 2016;(14):e1002585. doi: doi.org/10.1371/journal.pbio.1002585.
- Velten L, Haas SF, Raffel S, Blaszkiewicz S, Islam S, Hennig BP, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nature cell biology. 2017;19(4):271-81. doi: 10.1038/ncb3493. PubMed PMID: 28319093.
 (1 votes)
(1 votes)2 thoughts on “A glimpse of a dynamic cell fate decision”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
Very stimulating reading!
I would however “disagree” on what you described as “phenotypic plasticity of cells”, which is based mainly on the “transcriptomics” plasticity captured by single-cell RNAseq. You’ll agree that, functionally, cells are phenotypically very robust, in particular what we call “differentiated” cells (that’s why “we” function!). Whether the “observed” transcriptomics variability is a technical problem or an irrelevant “problem” for the cells, is still an open question. Does it make any difference (energetically) for the cell to produce 100 or 300 mRNA copies of a given gene? Even if it does, buffering at the protein level will make this irrelevant, as the ratio protein/mRNA is very high (see 10.7717/peerj.270). Because we are limited to “transcriptomics” and cannot do (yet) proteomics at single-cell level, we should be careful to prevent misleading generalizations.
And yes, stem cells are more “plastic”, transcriptionally and “phenotypically”, and that is what makes it very difficult to isolate these cells as a single “entity”. Your work contains some of the best experimental evidence for this unique stem cell features. The challenge is to understand how the transcriptional plasticity repeatedly found in stem/progenitor cells is central to their identity/function, and how is this “noise” generated/regulated? I guess that my question is whether stem cells know who they are, or whether they are an extreme case of multiple personality (dis)order?
You are indeed raising fundamental questions! Although we have no definite answer for them, I think that we can say a little more than what you suggest. It is absolutely clear now that the observed transcriptomic variability is real and not just a technical artefact. We are also know, that the proteomic variability is not less. Just consider a cytometry plot: the difference between the protein levels in cells of the same type may reach up to two or more logs! There are many different reports that demonstrate and quantify transcriptomic and proteomic variability. May I suggest you one of ours? http://journals.plos.org/plosone/article?id=10.1371/journal.pone.0115574
You can see that the protein levels may change several times during the same cell cycle!
A very interesting recent study has demonstrated (Lestas et al. Fundamental limits on the suppression of molecular fluctuations Nature 467, 174–178 (09 September 2010) doi:10.1038/nature09333) that the cell’s capacity to suppress these fluctuations is very limited. Since it is impossible to get rid of it, the “noise” is presumably part of the normal functioning. I think that the best interpretation of our observation is precisely this: external stimulations induce a kind of non-specific “ready to respond” activated state where the increased intracellular noise generates an essentially random gene expression pattern. This is indeed a kind of “multiple identity state”. The relaxation from this “activated” state to a less noisy stable phenotype is gradual and accompanied by slow fluctuations. A number of papers came to similar conclusion. I suggest two recent reports: http://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.1002585
http://journals.plos.org/plosbiology/article?id=10.1371/journal.pbio.2000640
The key question therefore is to understand the mechanisms of “noise modulation” as a basis of “plasticity”. However, I think that there are some conceptual issues need to be settled before we can consider cell plasticity: What is a cell phenotype? Can it be described by the transcriptome? By the proteome? By cell morphology? How long a phenotype has to be maintained to be considered as stable?