How do new cell types evolve? Sea urchins show the way….
Posted by ettenson, on 21 November 2019
We know surprisingly little about how evolution has created new cell types. One of the best examples of a recently evolved cell type comes from early sea urchin development. Most sea urchins produce a group of early embryonic cells known as micromeres- four small blastomeres that form by unequal cell division at the vegetal pole of the early embryo (Fig. 1).
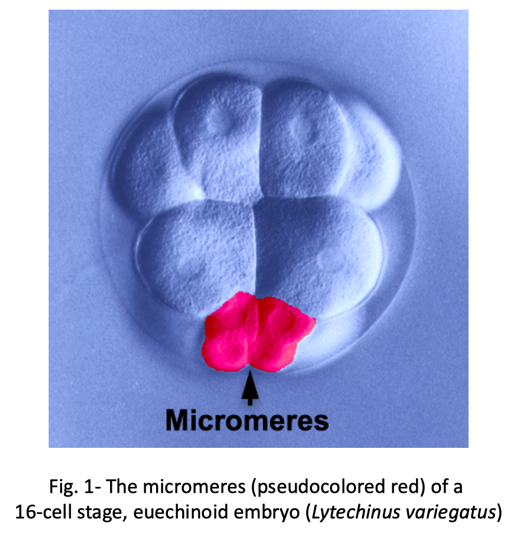
Micromeres give rise to primary mesenchyme cells (PMCs), a population of cells that moves into the blastocoel at the start of gastrulation and secretes a beautiful, biomineralized skeleton, which provides structural support for the larva. This is the developmental pattern seen in euechinoid sea urchins, a group that includes most species commonly used by researchers, and the pattern described in all developmental biology textbooks. Yet this developmental program is highly unusual within the echinoderm phylum. For example, the embryos of cidaroids, a small group of sea urchins that may be akin to “living fossils”, have variable numbers of micromeres and lack PMCs. In these embryos, mesenchyme cells migrate into the blastocoel and form a skeleton, but this occurs much later in development. A compelling body of evidence, including comparative studies in several echinoderm taxa, supports the view that the PMC lineage of euechinoids evolved relatively recently through the formation of micromeres and a heterochronic shift of an ancestral skeletogenic program into the early embryo.
Interestingly, like cidaroids, euechinoids produce late-migrating mesenchyme cells with skeleton-forming potential. These multi-potent cells, known as blastocoelar cells (BCs), ordinarily give rise to immunocyte-like cells. Their choice of cell fate is controlled by PMCs, which suppress the skeletogenic potential of BCs and direct them to instead to develop as immunocytes. The interaction between PMCs and BCs can be revealed by microsurgical experiments. If PMCs are ablated from the embryo, BCs undergo a striking change in phenotype; they adopt PMC-specific morphogenetic behaviors and secrete a correctly patterned skeleton. This switch in cell fate (a phenomenon that can be called “transfating”) is associated with the molecular reprogramming of BCs, which ectopically deploy a well-described skeletogenic gene regulatory network while extinguishing the expression of genes associated with an immunocyte fate. Remarkably, the effect of PMCs on BCs is titratable- the greater the number of PMCs that are removed from the embryo, the greater the number of BCs that transfate. The first indication of an interaction between PMCs and BCs came more than 50 years ago, but the molecular nature of the PMC-derived signal has remained a mystery.
In a recent study (Ettensohn CA, Adomako-Ankomah, A, 2019, The evolution of a new cell type was associated with competition for a signaling ligand. PLOS Biology 17: e3000460.), we discovered the elusive mechanism of the PMC-BC interaction. First, we showed through gene knockdown and inhibitor studies that an ectoderm-derived signaling ligand, VEGF3, and its cognate receptor, VEGFR-Ig10, are essential for the activation of the skeletogenic program by transfating BCs. Christian Gache’s laboratory first showed that these factors play an important role in PMC development and subsequent studies have revealed a conserved role for Vegf signaling in skeletogenesis throughout the phylum. One important gene activated by this signaling pathway in BCs is alx1, a gene that encodes a transcription factor with a pivotal role in controlling skeletogenic cell identity.
The requirement for VEGF signaling in BC transfating immediately suggested a possible mechanism for the PMC-BC interaction. Because PMCs express VEGFR-Ig10 from an early developmental stage, it seemed plausible that they might outcompete BCs for VEGF3, the ligand essential for activation of alx1 and downstream components of the skeletogenic gene network in BCs. This hypothesis led to two testable predictions. First, lowering the level of VEGFR-Ig10 expressed by PMCs should compromise their ability to suppress BC transfating. Second, over-expression of the ectoderm-derived ligand, VEGF3, should saturate receptors on the PMC surface and provide sufficient unbound ligand to induce the skeletogenic program in BCs, even in the presence of PMCs. Our study tested and confirmed both of these predictions, providing compelling support for the ligand-competition model (Fig. 2).

Of course, these results raise new questions. For example, we do not yet know the molecular connection between VEGF signaling and alx1 activation in BCs. Nevertheless, the findings are exciting for two reasons. First, to developmental biologists, they provide an example of the regulation of early embryonic cell fates by direct competition for a secreted signaling ligand, a developmental mechanism that has not been widely recognized. Second, to evolutionary biologists, they reveal that a novel cell type evolved by out-competing other embryonic cell lineages for an essential signaling ligand that regulates the expression of a transcription factor controlling cell identity. In the future, the micromere-PMC lineage will continue to be a powerful experimental model for elucidating developmental and evolutionary mechanisms that have led to the appearance of new cell types.
 (4 votes)
(4 votes)One thought on “How do new cell types evolve? Sea urchins show the way….”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
Was this new celltype additional to existing celltypes?
Or does it take the place of the previous celltype?