More Than a Powerhouse: How Mitochondria Help to Build an Embryo
Posted by Alexandra MacColl Garfinkel, on 16 October 2023
A recent paper was published in Developmental Cell, titled “Mitochondrial leak metabolism induces the Spemann-Mangold Organizer via Hif-1α in Xenopus”1. The researchers find that a mitochondrial membrane leak actives Hif-1α, which is sufficient to induce the Organizer and activate Wnt reporters. They propose that mitochondrial leak metabolism could be a general mechanism for activating Hif-1α and Wnt/β-catenin signalling. We caught up with the first author, Alexandra MacColl Garfinkel, PhD, to learn more about the story behind the paper.
How did you come to join the Khokha lab?
When I got to Yale University to start my PhD, I wanted to work in a setting that empowered independent and explorative science and used a well-established model and experimental framework. The Khokha lab provides such a wonderful space to do this. By utilizing available human congenital heart defect databases, and the well-established Xenopus model organism, students and post-docs can identify potentially important genes for human development and quickly screen for functional conservation in Xenopus. I was also deeply motivated by the underlying philosophy of Dr. Khokha’s lab, which is rooted in a commitment to provide discoveries made in the lab to physicians who work directly with families struggling with the realities of birth defects. Discoveries made in the lab can provide information to families about why this type of defect occurs and can inform genetic testing that is done before conception or for in vitro fertilization procedures.
Because most genes do not have well defined roles in development, this lab framework leads to an amazing diversity of focuses and discoveries. This can be challenging, as everyone in the lab is working on something different, learning as they go; but it’s also an incredible asset. Everyone uses the same tools to begin their projects, which means a lot of intra-lab collaboration. It also breeds an incredibly supportive learning environment. Everyone becomes a teacher in the lab, and lab meetings are exceptionally diverse and interesting. It is also an environment that encourages inter-lab and inter-departmental collaborations. As students and post-docs delve deeper into the molecular mechanisms of the gene, pathway, or cellular process they are studying, reaching out to experts to learn new techniques and theories becomes crucial! This really attracted me to the lab.
How did the project get started?
My project began with the study of the gene LRPPRC, identified in a patient with congenital heart defects. I quickly found data in the literature that patients with mutations in LRPPRC are often diagnosed with French Canadian Leigh Syndrome or related metabolic diseases. However, I also found that some of these patients have a variety of structural birth defects. LRPPRC is a known RNA binding protein critical for the maintenance of the electron transport chain (ETC) and ATP synthase. So, the metabolic phenotypes made sense. But based on the other patient data, I was interested in a possible role of LRPPRC in embryonic patterning.
By using the CRISPR/Cas9 gene editing system, I knocked down this gene in Xenopus and found a significant number of heart and craniofacial defects, as well as more severe neural tube patterning defects. The neural tube and broad patterning defects suggested an early role for lrpprc in development. I looked at patterning markers throughout development and eventually found that the earliest defects occur at gastrulation. These gastrulation defects are caused by abnormal mesoderm specification, specifically the expansion of the Spemann-Mangold Organizer. I was shocked. I had expected to find a loss-of-function phenotype, associated with the disruption of mitochondrial ETC and ATP synthase functions. I had assumed that mutating this gene would cause a loss of cell fate specification or even cell death due to a decrease in energy production or increase in autophagy. But instead, I found this incredible shift in mesodermal cell fate specification, suggesting there were metabolic mechanisms driving cell fate decisions.
What was known about the establishment of the Spemann-Mangold Organizer and the role of mitochondria metabolism in Organizer specification before your work?
The Organizer is an evolutionarily conserved tissue that is named for its ability to “organize” pluripotent embryonic stem cells into the structures of the primary body axis. In 1924, Hans Spemann and Hilde Mangold first identified the Organizer and showed its capacity to instruct the fate and organization of nearby cells. By transplanting a pigmented newt embryo “organizer” into a non-pigmented newt embryo, they were able to track the transplanted cells and confirm their capacity to organize the host-tissue into a second, ectopic, embryonic body.2
Since the advent of genetic cloning, a handful of signaling pathways have been investigated for their role in establishing the Organizer. In particular, the canonical Wnt/β-catenin signaling pathway stimulates the expression of key Organizer genes. β-catenin is stabilized in the dorsal region of the developing embryo. It drives Organizer gene expression there and eventually establishes dorsal and neural cell lineages. However, before the field of developmental biology exploded with genetic tools, developmental biologists in the early 20th century thought that metabolism could be involved in the Organizer’s function. This has mostly been forgotten, as the progression of developmental biology became increasingly defined by the identification of powerful genetic signaling cascades. Looking back though, it is interesting to see how even in the 1930’s and 1950’s this idea was percolating.
At the time, developmental biologists were trying to understand what signal could emanate from the Organizer to communicate with surrounding cells and instruct them so thoroughly. We now know that BMP inhibitors are expressed in the Organizer and are secreted to nearby cells. However, these early studies hit upon something intriguing. Although controversial due to the difficulty of reproducibility, some scientists including Norma Ornstein and John Gregg found that dorsal explants from amphibian embryos consume more oxygen than ventral explants3. There was a lot of speculation about how this might be related to signaling from the Organizer, but not about how this might relate to the establishment of the Organizer itself.
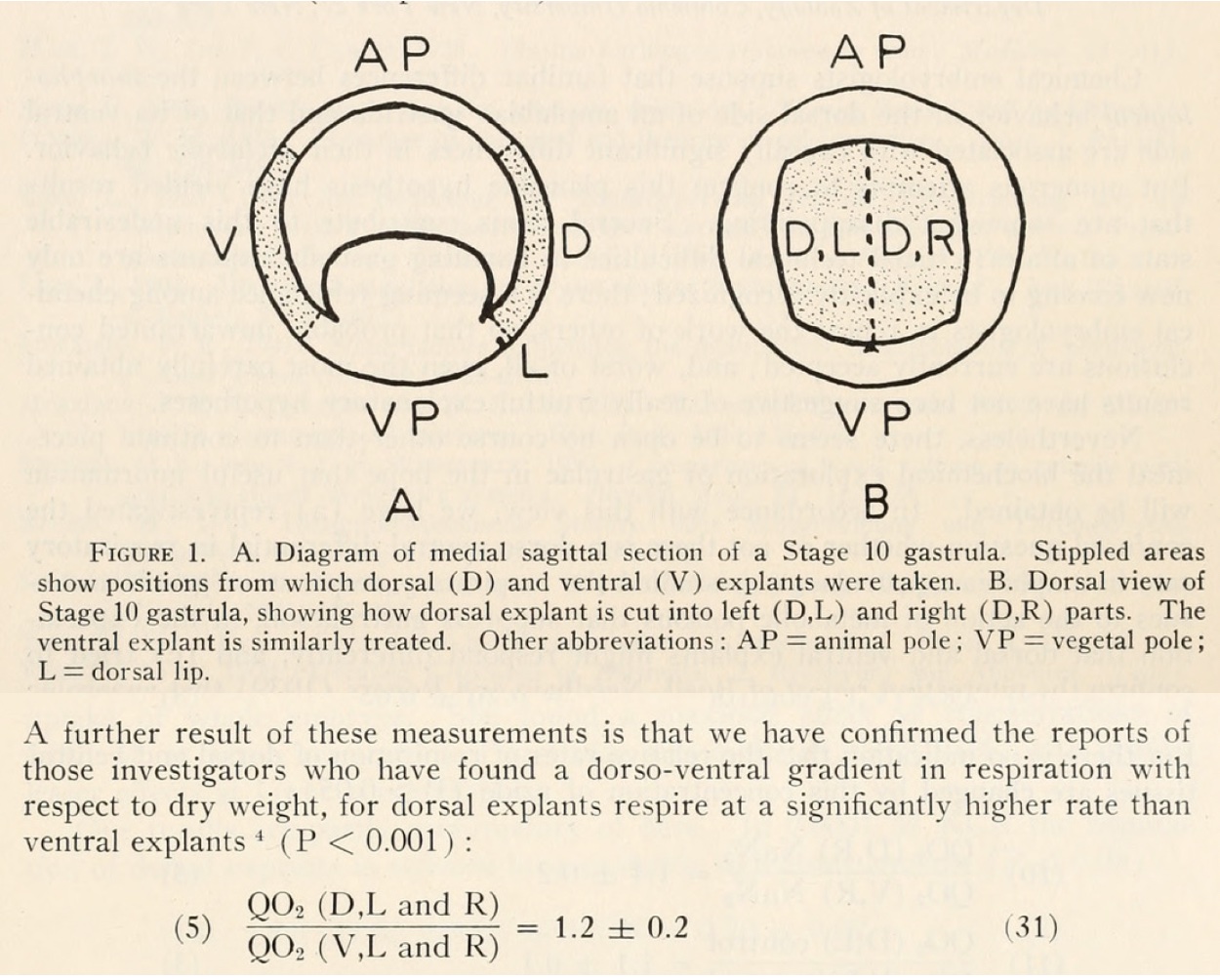
Can you summarize the paper’s key findings?
- Disrupting oxidative phosphorylation, by genetic or environmental manipulation (hypoxia, Oligomycin), results in the expansion of the Organizer by upregulating Hif-1α.
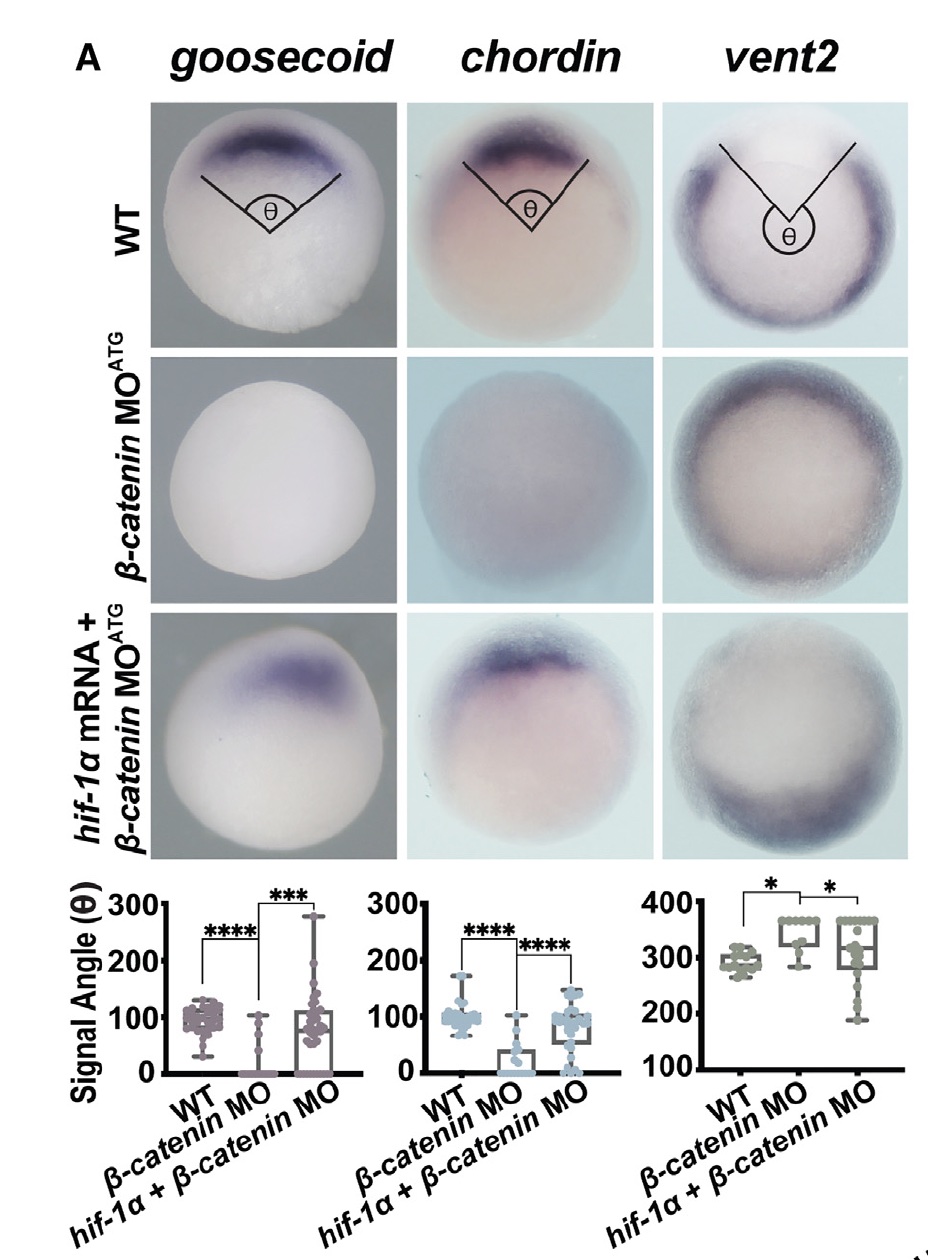
- Hif-1α overexpression leads to an expansion of the Organizer; knockdown of Hif-1α inhibits the expansion of the Organizer induced by mitochondrial manipulations or β-catenin overexpression.
- Hif-1α acts downstream from β-catenin to establish Organizer gene expression and can induce Organizer specification even in the context of β-catenin knockdown.
- Respiration in the Organizer is significantly higher than in the ventral mesoderm, suggesting Hif-1α is activated there despite functioning mitochondria. In fact, a functional ETC seems to be necessary for Organizer specification in wildtype embryos. This is due to the c-subunit of ATP Synthase acting as a mitochondrial membrane leak, which drives up ETC activity and increases oxygen consumption.
- Targeted expression of the c-subunit of ATP Synthase can induce an ectopic Organizer in any region of the embryo and can result in a second embryonic head and body axis. This effect is dependent on Hif-1α and occurs downstream of β-catenin.
Overall, our work suggests that mitochondrial leak metabolism could be a general mechanism for activating Hif-1α and Wnt/β-catenin-target gene expression. It is also incredibly cool to answer an almost 75-year-old question and finally know why the Organizer uses more oxygen than other tissues!
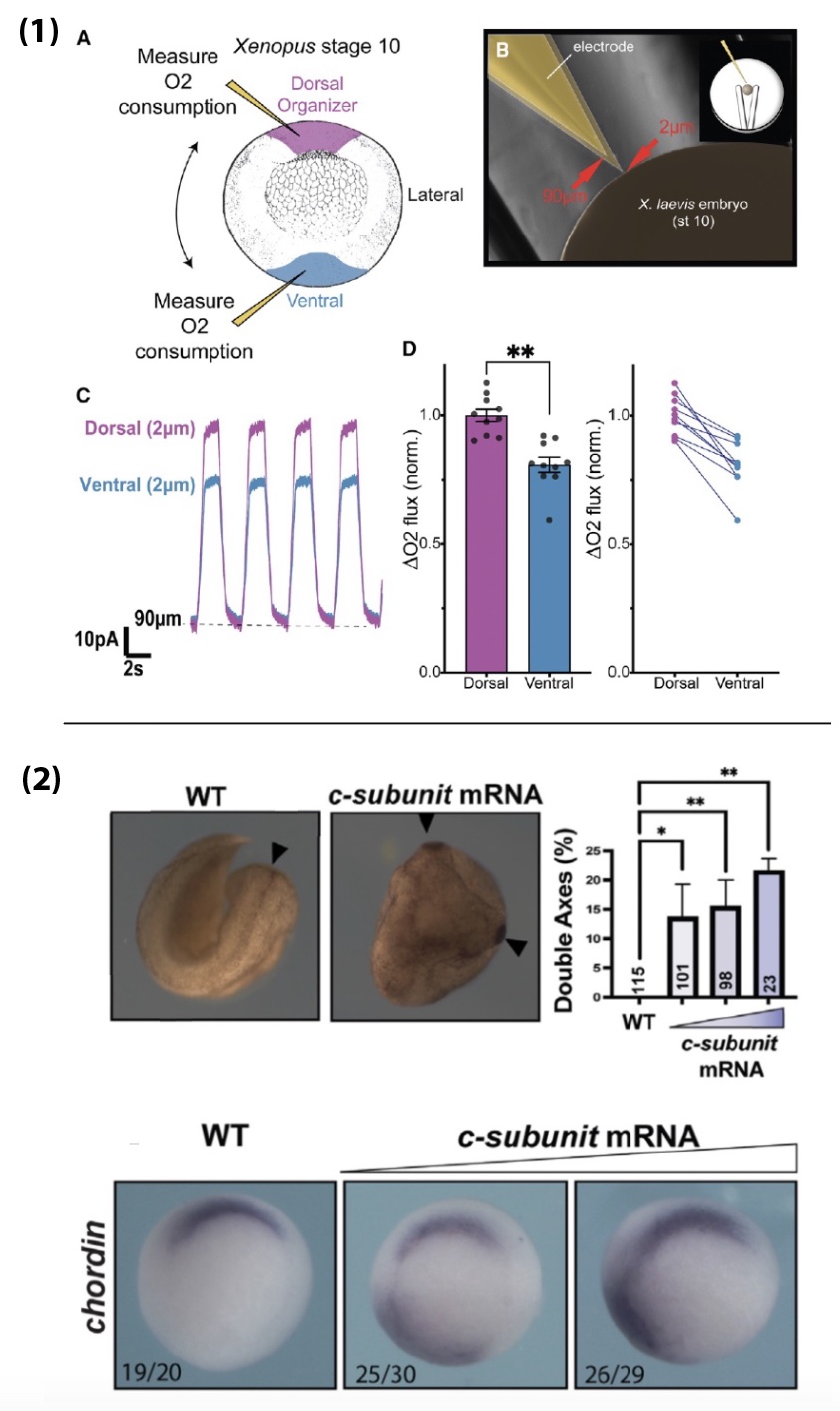
Were you surprised to find that Hif-1α is sufficient to expand the Organizer cell fates in normoxia?
When I first overexpressed Hif-1a, and saw such a clear expansion of the Organizer, I was stunned and relieved. It was a major puzzle piece falling into place. I had been trying to make sense of why β-catenin was not increased in lrpprc CRISPR mutants. When you lose β-catenin, you lose Organizer specification completely, and when you overexpress it, you get an expansion or duplication of the Organizer, so I was expecting β-catenin to be driving the phenotype in the mutants. Because of the role of Lrpprc in mitochondrial maintenance, I had made a list of possible metabolic proteins to manipulate to see if I could recapitulate this phenotype. With the Hypoxia data we obtained in collaboration with Dr. Andrea Wills at the University of Washington, and from helpful conversations with her, I started with Hif-1α and saved a lot of time!
In terms of being surprised about the normoxia situation – honestly, I think I was so focused on the data, and focused on getting to a point where I deeply trusted my findings, that it didn’t strike me as that odd. There is something magical about being a PhD student setting out into a new field, especially because you come in with very few pre-conceived notions. I read so much about Hif-1α in a range of contexts and from what I learned it seemed pretty reasonable that something could be going on to stabilize HIf-1a in the embryo. In the end, biology is what it is, whether we understand the nuances or not. In the case of Hif-1a, I trusted the data, and used the amazing work already published by others to start making sense of the results!
Can you postulate about how Hif-1a is activated?
I have a few ideas about this. Hif-1α is dynamically regulated by so many signaling pathways and molecules. Obviously, you have the oxygen piece, then there are other factors like a-Ketoglutarate vs Succinate/Fumarate, pyruvate, and lactate, that are involved in regulating the activity of the prolyl hydroxylase domain (PHD) proteins that target Hif-1α for degradation; there are genetic regulators and growth factor signaling pathways that interact with Hif-1α. Then there are the direct interactions that have been described between Hif-1α and β-catenin, TCF/LEF, YAP1, and more. I think that in the context of mitochondrial membrane leak, regional increases in lactate and pyruvate could result in their competitive binding to PHDs and inhibit Hif-1α degradation. It is also possible that the increase in oxygen consumption produces high ROS levels which can inhibit PHD activity. Additionally, I saw an increase in regional mRNA levels in the Organizer and dorsal ectoderm, suggesting possible regulation at the transcription and/or translation levels. We are currently exploring these possibilities and I am excited for what is to come.
Did you have any particular result or eureka moment that has stuck with you?
Oh my, I feel like there were so many. During this work, I often felt like I was in the dark, following the dim light of my data, which sometimes felt like it was taking me in circles. I had a LOT of opportunities to practice trusting my data, even though I couldn’t see where it was leading me. And then there would be these flares of light, from a new result, or from reading a paper, and something would click, and I would jump 10 steps ahead. The whole project was truly an incredible learning experience.
I know I should probably stick to one, but I really couldn’t choose between these!
- I have to say again, that the effect of overexpressing Hif-1α in the early embryo and getting such a striking Organizer phenotype was profound. It got us on track to begin understanding the mechanism. For me, it was the western blot I did to confirm Hif-1α is upregulated in lrpprc CRISPR mutants that really changed the game! That was when I did the experiments to confirm that losing Hif-1α rescues the lrpprc Organizer phenotype and I felt like I could begin sinking my teeth into the problem of what Hif-1α is doing early in development.
- Seeing that Hif-1α can rescue the Organizer in β-catenin depleted embryos – I was absolutely shocked.
- Going to London to perform regional Oxygen-consumption experiments in live embryos was such a privilege and a treat, then to find that the Organizer consumes more oxygen than other tissue blew me away. At the time I was such a novice in thinking about metabolism. Thank God for Dr Jonas and Dr Alavian – they laughed kind heartedly at my look of utter astonishment when we saw the result. They suspected what was going on immediately and helped me frame the concept of physiologically useful mitochondrial membrane leak. Seeing that the c-subunit leak could induce an ectopic body axis and Organizer was an incredible finale to this work.
And the flip-side: were there any moments of frustration or despair? What got you through?
Again, so many. Especially early on. I got the lrpprc ‘expanded Organizer’ phenotype and was so stumped because β-catenin wasn’t up. It took patience, creative experimenting, reading, and good conversations. When I read the 2017 papers that came out of Dr. Olivier Pourquié’s4 and Dr. Alexander Aulehla’s5 labs on glycolysis and cell fate specification during somitogenesis, it was huge. I finally felt like someone else was in the dark with me, shedding light on what could be going on. Even though our paper doesn’t focus on glycolysis, just being able to read about metabolism being involved in developmental cell fate and patterning was so helpful. Up until then, there was so little out there! Building out a new field of intersectional biology is difficult. It can feel lonely and frustrating, but also so exciting. I am deeply grateful for the people I’ve found along the way who have been exploring and defining the burgeoning field of developmental metabolism. For example, I just went to an EMBO Workshop at the EMBL in Heidelberg called “Developmental metabolism: Flows of energy, matter, and information.” It was incredible to see the diversity of backgrounds and fields represented by this excited group of people, all of them ready to collaborate to continue building out this field.
What is next for this story?
I am currently working with Dr. Jonas in the Endocrinology Department at Yale Medical School, delving deeper into the molecular mechanisms of how c-subunit, Hif-1α, and β-catenin collaborate to regulate embryonic patterning using mice and human stem cell models. I love this work and am so excited to continue learning about the mitochondria and metabolism side of things from Dr. Jonas. She has an incredible lab and skill set and I have learned so much from her already. In my own lab in the future, I hope to use my discoveries as a proof-of-concept to define other mechanisms that lie at the intersection of metabolism and known developmental pathways. I believe that exploring this intersection is crucial for the field developmental biology, but also for our understanding of how complex multicellular life evolved and diversified. I am so excited for what’s to come and to continue building and investigating the field of developmental metabolism!
Cited in this article:
1. MacColl Garfinkel, A., Mnatsakanyan, N., Patel, J.H., Wills, A.E., Shteyman, A., Smith, P.J.S., Alavian, K.N., Jonas, E.A., and Khokha, M.K. (2023). Mitochondrial leak metabolism induces the Spemann-Mangold Organizer via Hif-1α in Xenopus. Dev Cell. 10.1016/j.devcel.2023.08.015.
2. Spemann, H., and Mangold, H. (1924). über Induktion von Embryonalanlagen durch Implantation artfremder Organisatoren. Archiv für Mikroskopische Anatomie und Entwicklungsmechanik 100, 599-638. 10.1007/bf02108133.
3. Gregg, J.R., and Ornstein, N. (1952). Anaerobic Ammonia Production by Amphibian Gastrulae Explants. Biological Bulletin 102, 22-24. 10.2307/1538619.
4. Oginuma, M., Moncuquet, P., Xiong, F., Karoly, E., Chal, J., Guevorkian, K., and Pourquie, O. (2017). A Gradient of Glycolytic Activity Coordinates FGF and Wnt Signaling during Elongation of the Body Axis in Amniote Embryos. Dev Cell 40, 342-353 e310. 10.1016/j.devcel.2017.02.001.
5. Bulusu, V., Prior, N., Snaebjornsson, M.T., Kuehne, A., Sonnen, K.F., Kress, J., Stein, F., Schultz, C., Sauer, U., and Aulehla, A. (2017). Spatiotemporal Analysis of a Glycolytic Activity Gradient Linked to Mouse Embryo Mesoderm Development. Dev Cell 40, 331-341 e334. 10.1016/j.devcel.2017.01.015.
 (1 votes)
(1 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.