New genome reveals ancient toolkit
Posted by Brent Foster, on 23 July 2024
Hydractinia symbiolongicarpus is an emerging model to understand stem cell evolution
Stem cells can’t hide what they are. At least, that’s the takeaway from the newly sequenced genomes of two colonial hydroids, Hydractinia symbiolongicarpus and Hydractinia echinata. I recently sat down with Dr. Christine Schnitzler of UF’s Whitney Laboratory for Marine Bioscience to talk about her experience assembling these genomes — a huge collaboration that started pre-pandemic, slowed down during the pandemic, and finally culminated in a paper published in Genome Research in March. Our conversation covered topics spanning from the logistics of large collaborations (including the need to find the right people), the shared molecular vocabulary that stems from genome projects, and the scientific merit of studying emerging systems.
“All genome projects are huge,” said Schnitzler. “I would never have been able to do this myself.”
This particular project has been 10 years in the making. In that time, sequencing technologies and analyses kept changing. The research team opted to swap the Illumina short-read platform for Pac-Bio long-read sequencing to dig deeper into two unconventional model organisms: Hydractinia echinata and Hydractinia symbiolongicarpus.
The genomes of these two cnidarians had a few surprises. First, the two genomes were quite different in size — Hydractinia echinata was 775MB and Hydractinia symbiolongicarpus was 514MB. And that’s not just based on sequence. The research team isolated single cells, stained nuclei with propidium iodide and ran them through a flow cytometer against a known standard. Second, based on the researchers’ analysis, these two species’ genomes diverged around 19 million years ago. That may seem like a long time, but if you consider that two different strains of the same species of jellyfish are estimated to have diverged 45 million years ago, then a true species divergence 19 million years ago is surprisingly recent.
“I thought that was pretty cool,” said Schnitzler.
Genomes offer a common language for biologists to understand similarities (and, importantly, differences) between species. To that end, Schnitzler’s team took a lot of time and care into making this resource available for anybody with even a cursory interest. They also developed a web portal for both Hydractinia species that allows curious biologists to dive deeper into a very, very granular level of gene evolution between two closely related genomes. They also generated a single cell RNAseq browser.
“That’s kind of fun. The whole point is to make this genome accessible and useful to the community,” said Schnitzler. “It was a priority for us to build that resource, which was not easy,” she added with a laugh.
“Collaboration is key. And having the right people is very helpful.”
At the end of our discussion, I gave Schnitzler a chance to respond to the “Questions For The Author” from PreLighter Isabella Cisneros who spotlighted the preprint last September (check out the PreLight article here). I’ve included her questions, numbered 1–3, as well as Schnitzler’s responses below.
Questions For The Authors (from PreLighter Isabella Cisneros):
1. How do you reconcile the large number of shared i-cell marker genes with the higher proportions of phylum-specific and cnidarian-specific genes in the H. symbiolongicarpus genome?
We took the entire genome and ran an orthology clustering of all predicted proteins from 49 other animals, including 16 cnidarians. We put them into bins once we got the clusters back. Are these genes in a “multi-species orthogroup?” That means they group with genes from animals outside of cnidarians, so they’re shared more widely. A lot of the other categories were cnidarian-specific.
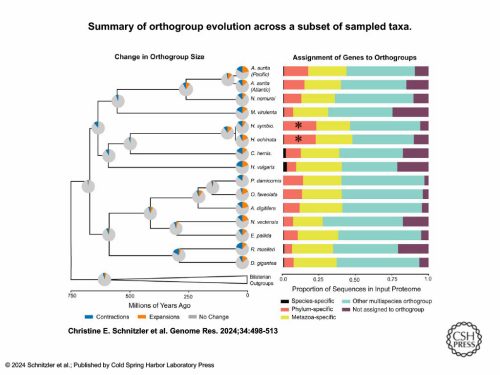
This graph shows just the cnidarians and a few other animal outgroups. Those two [asterisked] red bars are further out than all the other cnidarians — they had the highest proportion of genes in their genome that were specific to their phylum.
If you apply that same clustering to the most highly expressed genes that are specific to just the i-cells, we found that i-cell genes are mostly shared with other animals (that was the title of the paper).
How do I reconcile that? I think it means that stem cells are exactly what they are.
They’re undifferentiated cells that have to use basic cell characteristics — cell cycle genes, genes that help with proliferation, genes that are involved in maintaining stemness — and these are universal things that all animals have developed and retained throughout evolution.
That does not mean that stem cells of Hydractinia are exactly homologous to the stem cells of other animals. Cell types can evolve very quickly, but the underlying genes that make stem cells stem cells are very highly conserved.
There are a lot of cnidarian-specific genes in their genomes. It’s just in their stem cells, they’re not using those very much. But when you think about stem cells, they’re not really unique to cnidarians. All animals have some type of a stem cell — that’s just how it works.
2. Towards the end of the preprint, you claim that it remains unclear whether other animals share the same toolkit of genes, or whether these toolkits are instead partially overlapping. While further studies will be necessary to determine this, at this stage, what do you anticipate to be the case?
It’s very hard to relate cell types and say they have a shared evolutionary origin of the cell type. If you look at stem cells in Hydractinia and then look at stem cells in planarians, they have a lot of shared characteristics. But what about all the animals in between? Where did their stem cells go? I think that’s a very interesting question.
I think getting down to this core level of genes and gene regulatory networks that are controlling these types of cells, to me, might be more interesting and more informative from an evolutionary perspective than trying to absolutely say this cell type first arose at some point and then shifted. It’s not really about the cell types. It’s more about the core genes that are involved and finding out their function. And it’s really hard. I think the only way really is to drill down on function within several animals and then try to see if they relate to each other.
It’s good to look at informative positions on the tree, as a lot of people try to do, and try to gain as much insight as you can from looking at those different places. I don’t think there’s going to be 100% overlap. But I think there will be themes that emerge — categories of genes that may be similar. You could group them by orthology, but maybe not exactly by BLAST.
It’s a question for the future. I think people are trying to tackle it, but it’s going to take more than these genome-wide approaches. We’ll have to go back to the lab and do functional testing to get at those questions.
3. Given the different evolutionary trajectories that H. symbiolongicarpus and H. echinata have followed since divergence, what kinds of studies would be better suited for each species, if any? What could be gained by using both species in a comparative framework?
Most people have dropped H. echinata as a research organism. It’s got a bigger genome that’s not as well assembled. And no one’s maintaining them in the lab, anymore. They’re just harder to keep in culture long term. There may be some ecological and other questions that might be more interesting with echinata, and it would be cool if someone picks it up now that the genome is available.
On the other hand, H. symbiolongicarpus just grows and grows and grows indefinitely. After years and years of being in culture, we can still get them to spawn on a weekly basis and get tons of embryos. It also has a smaller genome that ended up with a better assembly.
Smaller genome, easier lab culture — all of that screams, “Work on this one!”
So for us, the path forward is symbiolongicarpus. Now we can talk and really understand each other when we’re talking about a particular gene or a particular process. Now we have this resource. It’s a starting point.
One interesting thing to think about is what is unique about Hydractinia biology? I like Hydractinia because it is different. It’s colonial with a polymorphism of polyp types. We have feeding polyps, sexual polyps, defensive polyps. Because we have this diversity just within a single colony, there are a lot more questions to ask. It’s a different type of development, a different kind of asexual reproduction.
The other thing that I think is super cool is its lifecycle. It’s a hydrozoan that has lost the medusa stage. So there’s no jellyfish stage. Its next closest sister group is Podocoryna carnea, which does have a polyp and a medusa stage. That genome is being sequenced now. So when we have these two genomes — Podocoryna and Hydractinia — you can now start to understand the difference between a hydrozoan genome that produces the medusa jellyfish and the genome that doesn’t. I think some of these comparative studies with new genomes are really, really exciting. And I think with unlimited resources, we would have perfectly assembled genomes for those two groups and start doing more experiments to try to understand the unique biology of Hydractinia.
One thing our paper does not really focus on is regeneration. Hydractinia symbiolongicarpus is a model of regeneration. There’s a huge amount of knowledge we can learn by studying regeneration in this animal. There’s been some seminal ground laying papers about regeneration in this model, but doing updated studies, which we have some data, hopefully soon we can update and talk more about how this animal achieves its amazing regenerative abilities.
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.