Temperature, variability and the robustness of development
Posted by Michael Dorrity, on 29 December 2023
A recent Cell paper finds that proteostasis governs differential temperature sensitivity across cell types in zebrafish embryos. First author Mike Dorrity tells us more about the story behind the paper.
How did you come to join Cole Trapnell’s lab and how did the project get started?
My PhD lab shared a work area with Cole’s in Seattle, so I spent a lot of time chatting with his group. One time, they were all heading out for pizza + drinks, and, being a graduate student, I tagged along hoping for an easy meal. As I was a bit out of place, some jokes were made about me being a new lab member, so Cole played along and asked: “Since you’re joining the lab, what are you going to work on?” He only seemed to be half-joking, so I had no choice but to give a half-serious response: “how temperature influences developmental robustness, something like that.” I believe that got the ball rolling. After discussing with other Trapnell lab folks working on fish, we hatched initial plans for temperature experiments.
What was known about how developing zebrafish embryos respond to temperature stress before your work?
Well, I have to split that one up a bit: (1) I think every zebrafish researcher knows something about how they respond to temperature, since slowing or speeding up development is practically very useful. Do you know what time you have to wake up to study 15-somite stage animals? The relationship between temperature and the rate of development is well-documented, and I refer to the chart in Kimmel et al 1995 constantly. (2) For temperature stress, the outcome is a bit different – there are developmental phenotypes that pop up over and over again. We knew about some of these phenotypes, such as a bend in the body axis, and, from previous work, even knew some of the molecular consequences of temperature stress: heat-shock protein induction, regulation of protein homeostasis.
Can you summarise the findings of the paper in one paragraph?
All in all, I think it can be summarized in one sentence: cell types do not respond equally to temperature. That sounds trivial, but it’s counterintuitive in the context of what we know about the heat shock response (uniform across cells) and of what we know about the acceleration of developmental rate at increased temperature (must be uniform-ish, as embryos are still viable). Basically, when you look closely at all cells at these sub heat-shock temperatures, you can see that temperature destabilizes the fidelity or robustness of development of certain cell types more than others. These cell type-specific sensitivities might not be so obvious when looking at the whole embryo under a stereoscope, but they explain the common failure modes of embryogenesis and the phenotypes that emerge as embryos approach the limits of developmental robustness.
Were you surprised to find that some cell types display accelerated developmental rates more than other cell types in response to the same elevated temperature?
Absolutely, this result completely changed how I thought of the problem. I come from a background informed by the biochemistry and molecular control of the heat shock response – the dogma there, rightfully so, is that all cells more or less mount the same response to stress: stop making proteins and prevent the existing ones from falling into misfolded, non-functional states. That makes good sense, but the embryo context is so different from something like yeast; different cells have drastically different functions and thus express a unique repertoire of proteins to carry out those functions. From that perspective, it’s not so surprising that cells respond differently. Beyond that, cells have many ways to sense temperature that can also vary in the embryo. For example, plasma membrane fluidity increases as a function of temperature, but this increased fluidity is also necessary for migratory cell types.
Can you describe how you captured phenotypic variability across individuals raised at different temperatures?
Yes! This technique is indebted to previous work in the Trapnell lab, [Srivatsan, McFaline-Figueroa, Ramani et al 2020] as well as efforts by Lauren and Sanjay in the “sister” publication to this one [Saunders, Srivatsan et al 2023]. Basically, the trick is: during embryo dissociations, we add barcoded, single-stranded DNA oligos and fix them to nuclei that come from a single embryo. This ensures that we can link all those cells back to the embryo-of-origin after our single-cell RNA-seq library preparation. Our phenotype in this case is actually whole-embryo cell composition, and you can only get measures of variability if you measure many individual fish (n > 100). This molecular trick unlocks individual-level data, and we can then perform statistics on the resulting cell counts, which would be otherwise be challenging to acquire for all cell types.
Can you postulate about the mechanisms through which the UPR controls temperature-induced developmental acceleration?
I would be happy to, and I encourage any potential reader to reach out if they’d like to discuss further, because this is an exciting new direction for us. The unfolded protein response, or the integrated stress response more broadly, has access to global control of protein synthesis in the cell. Under protein folding stress in the ER, UPR will put the brakes on translation. Control of translation rate (as well as protein degradation rate), has been linked to species-specific differences in the control of developmental rate, so I think any mechanism that feeds into protein synthesis rate has the potential to affect timing (we also saw slowdown of development when we inhibited translation). I think the sensing of protein concentration, protein synthesis, and protein folding in the ER is an efficient proxy for temperature. If you take away this sensing apparatus, the embryo doesn’t accelerate like it’s supposed to, despite being raised at a higher temperature.
Did you have any particular result or eureka moment that has stuck with you?
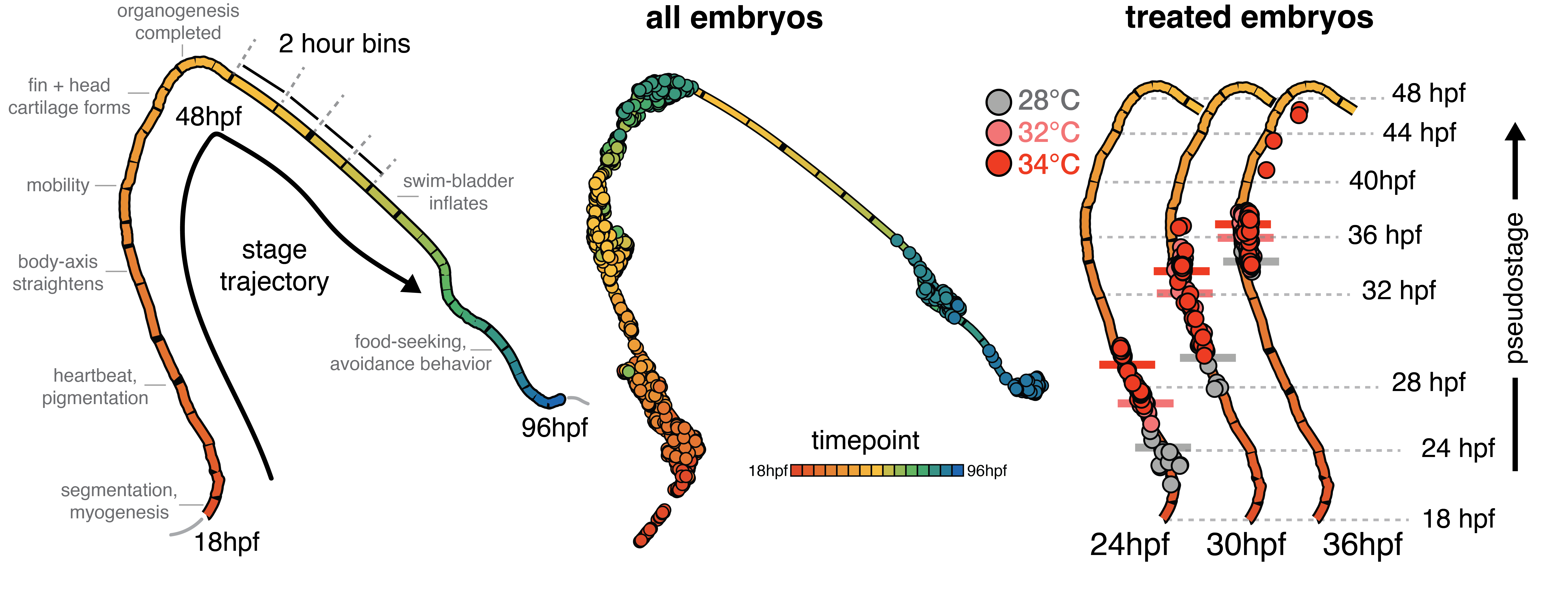
For me, it’s that an enormous amount of variation in the developmental-series scRNA-seq data is explained by the time or stage of the embryo. Again, this may sound trivial – of course gene expression patterns change over time! What has really stuck with me is how developmental timing, cell composition and transcription go hand-in-hand, even down to timescales of minutes (see Figure). Being immersed in the quantitative/statistical thinking of Cole’s lab made me realize how this simple observation can profoundly affects interpretation of your results; many factors (time, temperature, cell type..) are squabbling over the variation in your data, and it’s up to you to break them up and see what all the fuss is about.
And the flipside: were there any moments of frustration or despair?
Much of this project was completed between the peaks of the initial strain and Delta variant of SARS-CoV-2; despair was unavoidable. On the other hand, the constant questioning during those months was balanced by the task of being a scientist. I was very lucky to have co-authors with me at 2am in N95s, wearing headlamps (I cannot remember why) to dissociate embryos. The ensuing virtual meetings, discussions, optimizations, and laughs were essential in reminding me how science feels at its best: a community of people finding answers in the face of uncertainty.
What’s next for this story? And what’s next for you personally?
What’s next is: why are some cells more sensitive to temperature? Is this at all meaningful in the evolutionary context of temperature adaptation? Thankfully, I just started my group at the EMBL (https://www.dorrity-lab.com/) in Heidelberg, Germany surrounded by amazing colleagues and fellows to pursue these questions. If these questions sound exciting to you, reach out!
 (No Ratings Yet)
(No Ratings Yet)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.