The Tails of Fate
Posted by Ahmed Elewa, on 18 January 2018
The epic journey of embryogenesis begins with a set of maternal instructions. These instructions are in the form of transcribed mRNA, some even translated into proteins and ready for action. However, many of the critical maternal mRNAs are inactive and must be delivered to the right cell and activated at the right time to encode proteins necessary for development. Regulation of inactive mRNA transcripts is largely dependent on RNA binding proteins (RBPs) (1). However, the mechanisms whereby RBPs reactivate maternal mRNAs in the early embryo are largely unknown. As a doctoral student in Craig Mello’s lab, I was interested in how some RBPs regulate maternal mRNAs responsible for determining the developmental fate of embryonic cells. I wondered how these RBPs turn some cells into muscle and others into neurons, for example.
To pursue this quest, I used the nematode C. elegans, a powerful genetic model to discover genes involved in embryogenesis and to study how they interact together. Mutations in several RBP-encoding genes result in dead C. elegans embryos with cell fate errors, such as germ-cells turning into gut, or neurons becoming muscle (2-4). This further highlights the importance of RBPs in cell fate determination. I was especially fascinated by POS-1, an RBP present in posterior cells of early embryos (5). Depletion of POS-1 results in embryonic death and a complete loss of intestine (gut) (Figure 1, pos-1 mutant). I set out to study the role of POS-1 in the regulation of maternal mRNAs critical for gut fate.
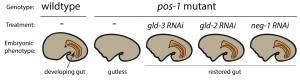
To identify the mechanism whereby POS-1 specifies gut fate I conducted an RNAi suppressor screen. I silenced hundreds of genes and asked whether gut fate in POS-1 deficient embryos could be restored. Interestingly, silencing another RBP-encoding gene, gld-3, restored gut in pos-1 mutant embryos (Figure 1, gld-3 RNAi). This result showed that RBPs can positively (POS-1) or negatively (GLD-3) affect gut fate.
Previous studies had shown that GLD-3 recruits the cytoplasmic poly(A) polymerase GLD-2 to mRNAs (6). GLD-2 then adds adenosine (A) nucleotides to mRNA tails to activate their translation to protein. Consistent with GLD-3 and GLD-2 functioning together, silencing gld-2 also restores gut in POS-1 deficient embryos (Figure 1, gld-2 RNAi). This suggested that both GLD-2 and GLD-3, by elongating poly(A) tails, were involved in POS-1 regulation of gut fate specification. Therefore, I asked which maternal transcripts being activated by GLD-2 and GLD-3 were relevant for gut fate?
Among the genes that I discovered as regulators of gut development in pos-1 mutants was the putative transcription factor neg-1 (Figure 1, neg-1 RNAi). Remarkably, silencing GLD-2 or GLD-3 decreased the length of neg-1 poly(A) tails, resulting in neg-1 inactivation. This suggested that GLD-2 and GLD-3 could prevent gut formation through neg-1 activation. Pursuing neg-1 was risky, however, since it had no homologs and therefore nothing hinting at its potential function. During a lab meeting, I expressed reluctance to study neg-1 with the excuse that “there is nothing known about it, nothing, and no homologues!”. To which a postdoc retorted, “We spend our careers looking for something new instead of more of the same. You have a shot at something novel and you’re throwing it away!” It was then that I realised we were before true novelty, for it was even beyond my own preconceived impressions of what qualifies as new.
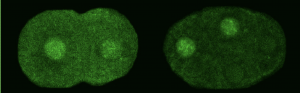
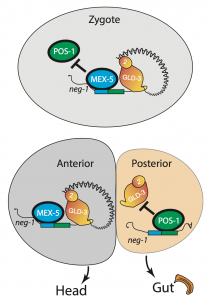
We asked whether POS-1 actually specifies gut fate by inactivating neg-1. We hypothesized that POS-1 inhibits GLD-2/GLD-3 polyadenylation of neg-1, leading to gut fate specification. Consistent with this hypothesis, we found that silencing pos-1 increases the length of neg-1 poly(A) tails and consequently NEG-1 protein levels. Moreover, tagging NEG-1 protein with GFP showed that it was absent in the posterior cells where POS-1 is present after the two-cell stage (Figure 2). Discovering that NEG-1 was an asymmetric protein was the moment the entire project started making sense and the pursuit of a novel gene paid off (See Video below for the Eureka! moment). Most importantly, we also discovered that POS-1 can bind regions in neg-1 mRNAs near the poly(A) tails (Figure 3, Posterior). Taken together, our results showed that POS-1 promotes gut fate in the posterior by preventing GLD-2/GLD-3 activation of neg-1 mRNA (7).
Considering the dramatic effect of gld-2 or gld-3 knockdown in pos-1 mutants (complete restoration of gut) (see Figure 1), I then asked if cytoplasmic polyadenylation was a general process in the early embryo rather than a neg-1 specific affair. We assayed poly(A) tails of thousands of transcripts following GLD-2 or GLD-3 depletion using a method based on next-generation sequencing (PAT-seq). I found that maternal mRNAs of hundreds of genes are polyadenylated by GLD-2 and GLD-3 and that some, like neg-1, are regulators of cell fate. In the future it would be exciting to explore the roles these GLD-2/GLD-3 activated maternal mRNAs.
One critical question remained: If POS-1 represses NEG-1, how could both proteins be present in the zygote? I noticed that one of the POS-1 binding sites in neg-1 mRNAs overlapped with a region recognized by another protein called MEX-5. POS-1 and MEX-5 belong to the same group of RNA binding zinc-finger proteins. MEX-5 is also critical for cell fate determination, but unlike POS-1 is present mostly in anterior cells of the early embryo (Figure 3, Anterior) (3). In the zygote, however, MEX-5 and POS-1 are present in the same cell. We found that MEX-5 outcompetes POS-1 in binding to neg-1 mRNA in vitro. Moreover, translation of neg-1 mRNA to protein required MEX-5. This suggested that MEX-5 enables the activation of neg-1 transcripts by blocking POS-1 binding. An intricate circuit was emerging before us: POS-1 represses neg-1 in the posterior to protect gut fate. Conversely, MEX-5 liberates neg-1 from POS-1 repression in the zygote and future anterior cells so that neg-1 can in turn repress gut fate where the head must develop (Figure 3). Morever, the PAT-seq analysis revealed that POS-1 represses mex-5 mRNA as well, consolidating the anterior/posterior divide.
In the perennial quest to discover the logic of life, the embryo has been the tome of reference. My doctoral research in the Mello lab has contributed to this long tradition of biological studies by linking a family of RNA binding proteins (CCCH zinc-fingers) with cytoplasmic polyadenylation and demonstrating the importance of this connection during embryogenesis. Cytoplasmic polyadenylation is prevalent throughout the tree of life. It is a crucial, for example, to quickly activate mRNAs and produce synaptic proteins in mammals during learning (8). CCCH zinc-fingers are also deeply rooted in life, from single-cell organisms to humans. These RBPs have been shown to mediate the swift removal of mRNAs, for example, in the immune system during acute inflammation (9). My doctoral research has shown that these conserved mRNA regulators collaborate at the commencement of nascent life to decipher the cache of maternal instructions for cell fate and beyond.
The epic journey continues, for the tales of fate do not end here.
References:
- L. Weill, E. Belloc, F. A. Bava, R. Mendez, Translational control by changes in poly(A) tail length: recycling mRNAs. Nat Struct Mol Biol 19, 577-585 (2012)
- C. C. Mello, B. W. Draper, M. Krause, H. Weintraub, J. R. Priess, The pie-1 and mex-1 genes and maternal control of blastomere identity in early C. elegans embryos. Cell 70, 163-176 (1992)
- C. M. Schubert, R. Lin, C. J. de Vries, R. H. Plasterk, J. R. Priess, MEX-5 and MEX-6 function to establish soma/germline asymmetry in early C. elegans embryos. Mol Cell 5, 671-682 (2000)
- B. W. Draper, C. C. Mello, B. Bowerman, J. Hardin, J. R. Priess, MEX-3 is a KH domain protein that regulates blastomere identity in early C. elegans embryos. Cell 87, 205-216 (1996)
- H. Tabara, R. J. Hill, C. C. Mello, J. R. Priess, Y. Kohara, pos-1 encodes a cytoplasmic zinc-finger protein essential for germline specification in C. elegans. Development 126, 1-11 (1999)
- L. Wang, C. R. Eckmann, L. C. Kadyk, M. Wickens, J. Kimble, A regulatory cytoplasmic poly(A) polymerase in Caenorhabditis elegans. Nature 419, 312-316 (2002)
- A. Elewa, M. Shirayama, E. Kaymak, P. F. Harrison, D. R. Powell, Z. Du, C. D. Chute, H. Woolf, D. Yi, T. Ishidate, J. Srinivasan, Z. Bao, T. H. Beilharz, S. P. Ryder, C. C. Mello, POS-1 Promotes Endo-mesoderm Development by Inhibiting the Cytoplasmic Polyadenylation of neg-1 mRNA. Developmental cell 34, 108-118 (2015)
- M. Ivshina, P. Lasko, J. D. Richter, Cytoplasmic polyadenylation element binding proteins in development, health, and disease. Annual review of cell and developmental biology 30, 393-415 (2014)
- S. A. Brooks, P. J. Blackshear, Tristetraprolin (TTP): interactions with mRNA and proteins, and current thoughts on mechanisms of action. Biochimica et biophysica acta 1829, 666-679 (2013)
 (2 votes)
(2 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.