Unraveling tissue interactions coordinating neuromuscular morphogenesis: a journey through serendipity
Posted by Francoise HELMBACHER, on 20 June 2018
An emerging trend in developmental biology focuses on the role of cell adhesion in modulating tissue morphogenesis. Spectacular advances have shed light on how modulation of adhesion between equivalent cells orchestrates the acquisition of forms. However, how interactions at complex interfaces between distinct neighboring cell types influence tissue growth remains to be elucidated. In a recent paper, staring mostly at a flat muscle that spreads subcutaneously and its partner motor neurons, I investigated how neuromuscular morphogenesis involves complementary activities of the Fat1 Cadherin in three interdependent cell types – myogenic cells, motor neurons, and the connective tissue through which muscles spreads.
Tissue-specific activities of the Fat1 Cadherin cooperate to control neuromuscular morphogenesis Françoise Helmbacher, PLoS Biology, May 16, 2018 | doi: 10.1371/journal.pbio.2004734 | PMID : 29768404
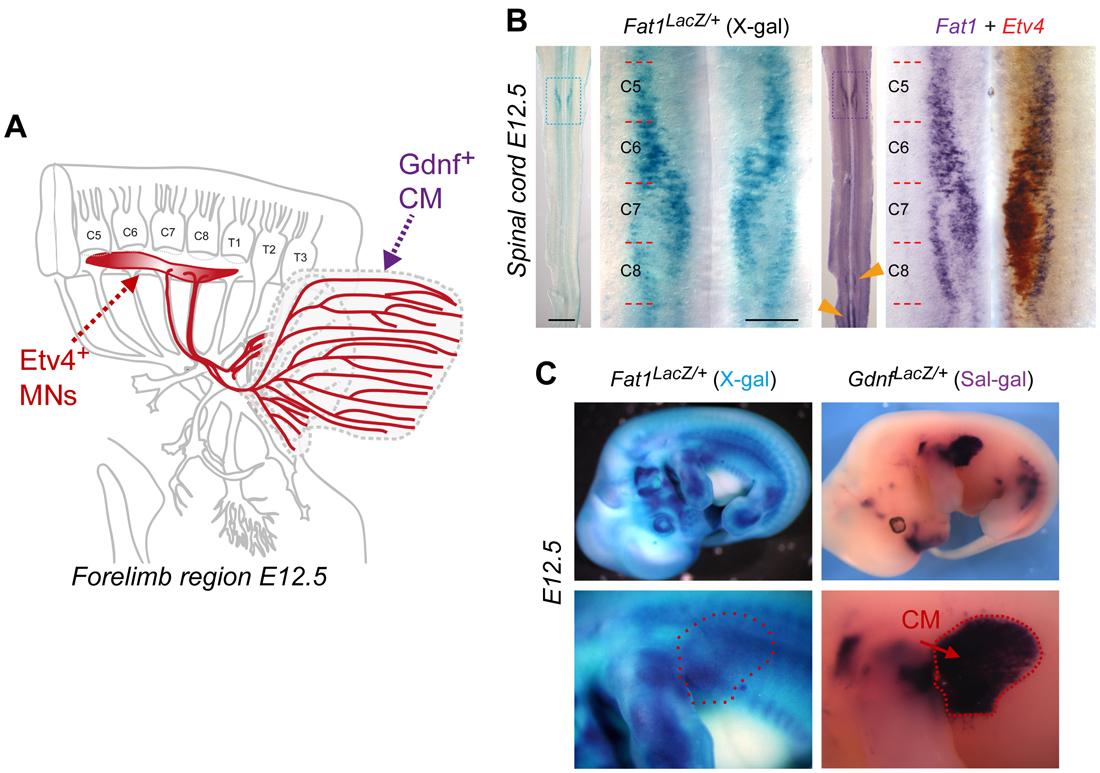
The story started as I was transitioning from being a post-doc in the lab of Rüdiger Klein in Munich, to establishing my own group in Marseilles. At the time, I was interested in events of communication between motor neuron (MN) populations, whereby a pool of MNs influences the specification of neighbor MNs. I was looking for molecules mediating such function, and Fat1 originally came up as one of my candidate genes. Although Fat1 met many of my criteria, it was initially considered a low priority candidate for the specific question I was asking (which I won’t discuss here). However, there were also good reasons for deciding to work on Fat1, aside from the main project of my freshly established lab: (1) Fat1 was expressed in my favorite MN population (Figure 1), a group of neurons recognizable by the transcription factor they expressed, which innervates a fascinating muscle, the Cutaneous Maximus (CM), (2) there were mouse mutants available (which by chance happened to be already in my former lab), and (3) through pioneer experiments with these mutants, I had gathered preliminary data indicating that Fat1 disruption resulted in defective specification of the CM motor pool and in defective axonal arborization within the target muscle – the phenotype I was expecting.
Whatever the underlying gene function, starting a project with a robust in vivo phenotype guaranteed that an interesting story would emerge from it, even though this was not my main project. For this reason, I kept this as my own side project. I had no hypothesis, but it felt like starting a classical forward genetics approach, with the advantage that I already knew the gene identity. This is also when the field of planar cell polarity (PCP) started blooming, mostly through work in Drosophila. The Drosophila Fat Cadherin, initially regarded as a putative tumor suppressor, had been attributed the mysterious function of propagating information on cell polarity within the plane of epithelia across long distances. I was fascinated by these studies, and curious to ask if MNs could propagate some polarity information to their neighbors too. Well, the story took a completely different path!
The two first key results that I had obtained were that 1) axons of the motor neuron pool innervating my favorite muscle, the CM, were clearly shorter in Fat1-deficient embryos, when all other nerves appeared unaffected; and 2) expression of one of the markers specifically characterizing this MN population was drastically reduced. Given the fact that at that time, I had selected Fat1 because of its selective expression in subsets of MNs encompassing the CM-innervating pool (marked by Etv4 expression, Figure 1B), these data were compatible with the simplistic and straight forward hypothesis of a cell autonomous activity of Fat1 in MNs. Thus, it looked as if this project would be devoted to finding out whether specification or axonal growth was affected first, and to tackle the mechanism, keeping PCP principles in mind, possibly using a combination of genetics, in vitro, and in vitro approaches.
–
Prior to jumping to the conclusion that Fat1 indeed controlled MN specification and axon patterning cell autonomously, it was essential to determine whether the target muscle, the CM, had developed normally in Fat1 deficient embryos. Any muscle defect might indeed represent a potential cause – or consequence – of these MN phenotypes, or might even indicate an independent phenotype. With my team, we therefore performed the experiment that changed the course of this story. We visualized muscle development by whole mount in situ hybridization to detect MyoD RNA in myogenic progenitors. To my surprise, this experiment turned out not to yield the anticipated negative result. Instead, Fat1-deficient embryos exhibited a series of very robust muscle defects affecting morphogenesis of subsets of muscles in the face and shoulder, phenotypes that we described in our PLoS Genetics paper in 2013 [1].
When we first saw these muscle phenotypes, there was no reason a priori to dissociate our findings on the altered development of a selective group of muscles and associated defects in the corresponding motor neuron population. However, as I was reflecting on the possible significance of these results in terms of health and biomedical implications, and as, in these early days, I was also in a phase of heavy grant writing, I wondered whether these phenotypes could possibly correspond to symptoms of a rare human pathology that would not have been assigned yet any genetic culprit. I thus started exploring the human genome to see where the human FAT1 gene was located, and discovered the OMIM database (Online Mendelian Inheritance in Man). It didn’t take long to realize that the 4q35 region where FAT1 was localized was also known for being subject to chromosomal abnormalities associated with a human muscular dystrophy called Facioscapulohumeral dystrophy (FSHD) [2]. I was really amazed to learn that the muscle wasting symptoms in this disease affected a highly regionalized group of muscles, with a topography reminiscent of what we had seen in Fat1-deficient embryos. I was also intrigued by the fact that additional non-muscular symptoms of this disease, auditory and vascular retinal abnormalities, were emerging as hallmarks of defective planar cell polarity.
At that time, this was a completely unexpected finding. First, the FAT1 gene was relatively far from the true FSHD locus (where changes in the D4Z4 macrosatellite repeat array were considered to be the primary event linked to FSHD). This distance (~3Mb) was sufficiently large to guarantee unambiguous genetic mapping of a disease locus, but was small enough for a genetic linkage, and for FAT1 regulation to be affected secondarily to chromatin architecture changes occurring as a result of the FSHD abnormalities (although the concept of topologically associated domains (TADs) was not as popular then as it has become). Even though a possible deregulation/contribution of neighboring genes had been considered, FAT1 had largely been ignored or dismissed. Nothing in the literature at that time indicated any likely involvement of Fat1 in muscle biology, whereas Fat1 disruption in mice had instead been reported to result in severe kidney abnormalities [3], the latter not being part of the panoply of FSHD symptoms.
This is when I decided to radically change the topic of the lab. I therefore sought to identify potential collaborators who would help approaching the human disease link. I was lucky to turn to Nicolas Levy, who headed a department of medical genetics at La Timone Hospital in Marseilles, and was really enthusiastic about this new idea. With his help, we were later joined by Marc Bartoli, who started a group on translational myology in Marseilles, and by Julie Dumonceaux (now at UCL, London). To make a long story short, this fruitful collaboration has led to date to 3 papers exploring the possible link between FAT1 dysfunction and FSHD [1, 4, 5]. First, my team provided compelling data indicating that disrupting Fat1 in mice caused muscular and non-muscular phenotypes with an FSHD-like topography [1]. Second, we detected lowered FAT1 RNA and protein levels in affected muscles from FSHD patients with classical diagnosis, at fetal [1] and adult [5] stages. Third, we identified FSHD-like patients carrying pathogenic FAT1 variants, in absence of the traditional FSHD-causal abnormalities [1, 4]. These FAT1 variants included single nucleotide variants affecting RNA splicing or amino-acid structure [4], but also copy number variants deleting a putative cis-regulatory element in the FAT1 locus [1]. Such deletions had the potential to cause tissue-specific depletion of FAT1 expression, thus offering a logical explanation for why some phenotypes resulting from Fat1 deficiency in mice were not part of the clinical picture of FSHD symptoms in human patients.
Because the FAT1/FSHD link was so unanticipated and provocative, the road to our first publication turned into a long and emotionally agitated roller coaster ride. As a result, this paper was submitted multiple times, and went through several rounds of revisions for several journals, until it proudly landed in PLoS Genetics in 2013 [1]. As in the meantime we had produced mice with a conditional Fat1 allele, we were able to show in this first study that Fat1 deletion in the myogenic lineage reproduced in part the aberrant dispersion of myogenic cells in the limb observed in constitutive knockouts [1]. I already knew however that this only gave a partial account of the complexity of Fat1 functions. I just figured that it was unnecessarily challenging to simultaneously report the identification of a potential new genetic actor in a human pathology (even when only considered a modifier gene) and the complexity of its biology. This deserved to be studied as a developmental question per se, independently of its relevance for this disease, which somehow is still a controversial issue, despite our genetic evidence obtained with the Levy/Bartoli groups.
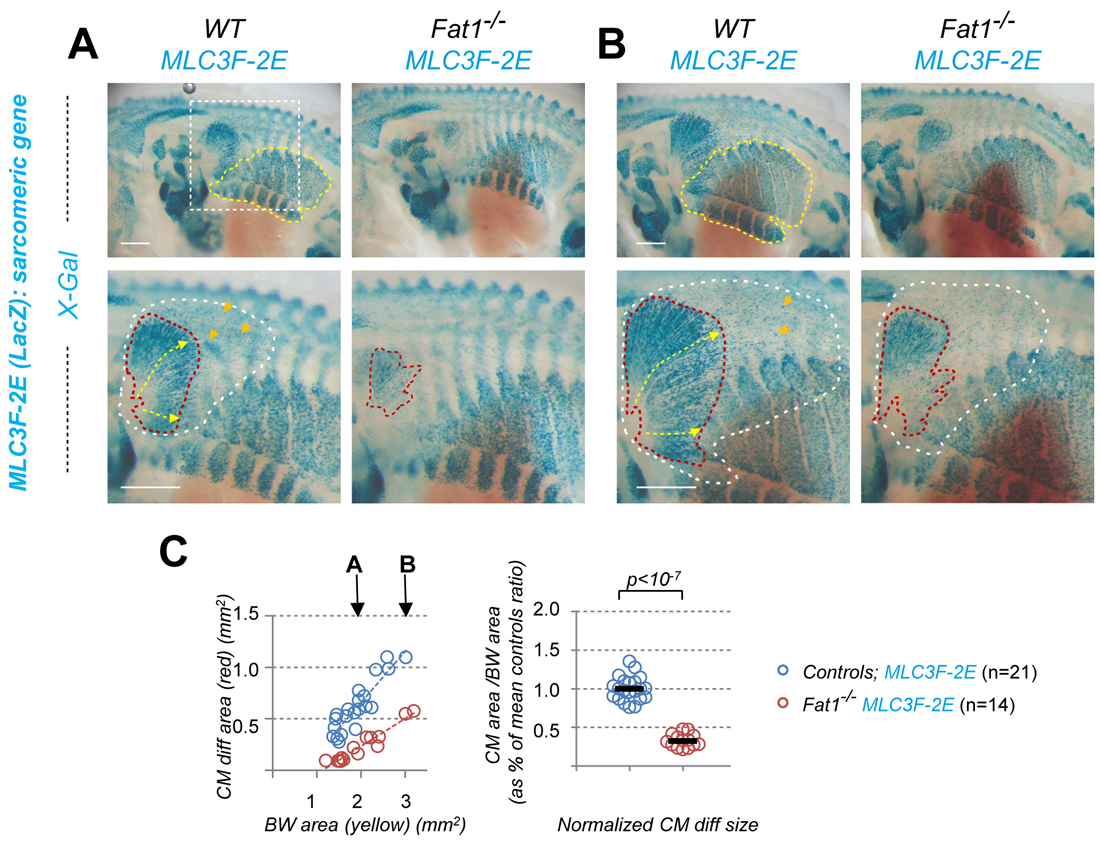
To approach the complexity of the Fat1-driven control of neuromuscular morphogenetic, the new study I have just published [6] gives a central position to one emblematic couple: the Cutaneous Maximus (CM) muscle, and its cognate MN pool. This MN pool can be recognized through its expression of the transcription factor Etv4, which is essential to specify the cell body position and dendritic architecture of these MNs [7]. The CM muscle emerges from the bulk of forelimb levels myogenic progenitors that undergo long range migration. Unlike other limb muscles, the CM muscle progenitors avoid entering the limb bud. Instead, they undergo a sharp caudal turn and start their subcutaneous progression. Throughout this migration phase, CM progenitors secrete Glial-cell-derived-neurotrophic-factor (GDNF), previously known for its neurotrophic activity and for its roles in motor axon guidance and in CM motor pool specification (as it induces Etv4 expression) [8]. Thus, by way of GDNF production, there is an intimate link between muscle progression and partner MN development. Among the muscles vulnerable to Fat1 deficiency, the CM muscle was also the easiest to approach quantitatively, by measuring the orientation of individual progenitor cells in our first study [1], and with simple area measurements in the new study [6]. Such an analysis illustrates how Fat1 disruption robustly interferes with expansion of the area occupied by Gdnf-expressing myogenic progenitors, by differentiating muscle fibers (Figure 2), as well as by axons of the cognate MNs.
–
Given my initial selection of Fat1 for its expression in the pool of MNs innervating the CM muscle, this was a region of non-myogenic Fat1 expression of obvious relevance to the phenotype. I was really excited by the possibility that Fat1 expression in the CM pool of MNs might represent a key driver of CM subcutaneous morphogenesis. Quantifications of morphometric parameters confirmed that MN-specific Fat1 ablation interfered with axonal growth and specification of the CM-innervating MNs. In addition, this also non-autonomously slows the subcutaneous progression of CM myogenic progenitors, hence expansion of the GDNF producing domain, without altering the rate of myogenic differentiation. However, this role turned out to be way more modest than what I initially anticipated, with an effect size considerably smaller than the phenotypes of constitutive mutants. This implied that another site of Fat1 activity had to be making a major contribution to neuromuscular morphogenesis.
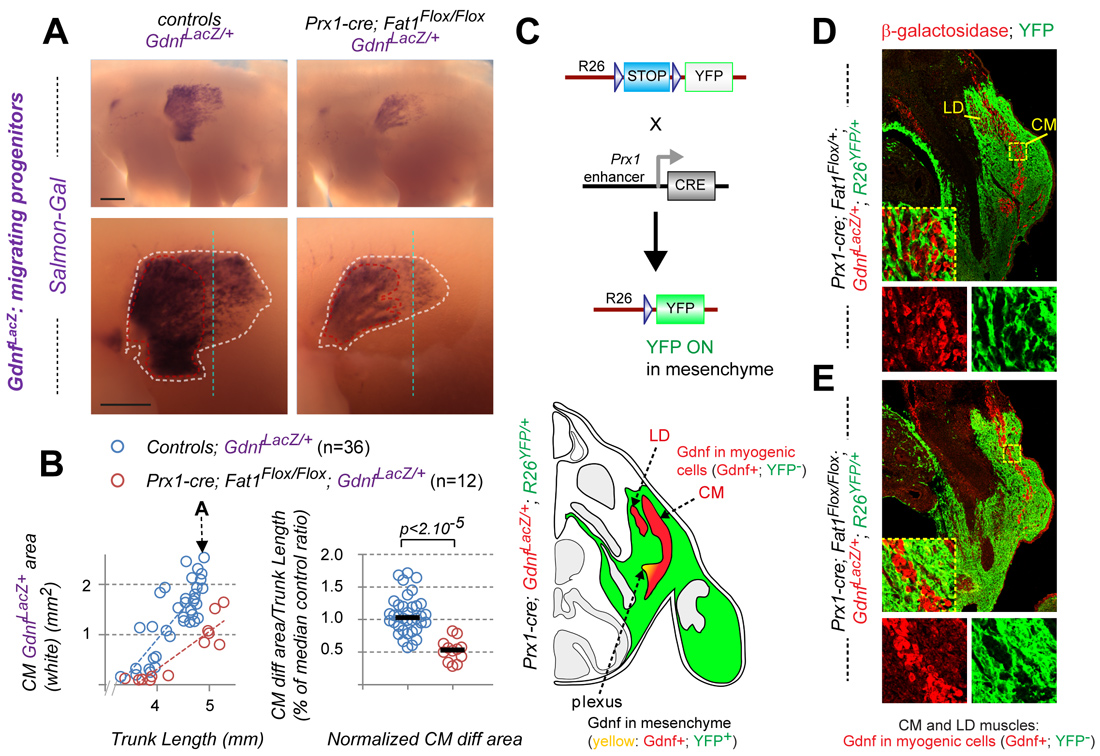
An unanticipated third site of Fat1 expression that turned out to play a major role is the loose connective tissue (CT) that surrounds muscles. During their collective progression, myogenic progenitors and motor axons explore such connective tissues and select their migration trajectory. The CM progenitors in particular encounter an increasing gradient of Fat1 expression in this tissue. Furthermore, Fat1 expression appears fully preserved in mouse mutants lacking migratory muscles (Met or Pax3 mutants, in which myoblast migration is abrogated), indicating that the main site of peripheral Fat1 expression represented non-myogenic cells. To establish the contribution of CT-Fat1 to muscle morphogenesis, Fat1 activity was ablated in the lateral plate derived mesenchyme, in a domain (driven by Prx1-cre) including the limb and flank connective tissues encompassing a large part of the territory through which the CM migrates. This led to severe non-cell autonomous disruption not only of the progression of CM progenitors and subsequent myofiber elongation, but also of motor axon elongation across the same territory, and of acquisition of CM fate characteristics. Similar results were obtained with a Cre line allowing stage-controlled Tamoxifen-inducible Fat1 excision in Pdgfrα-expressing connective tissue at the time of myoblast migration, with a lesser effect size matching the lower excision efficiency. These data identify connective tissue as a tissue type in which Fat1 activity is mandatory for neuromuscular morphogenesis. They imply that Fat1 signaling activity in CT cells is necessary for them to emit proper signals promoting collective myoblast migration, myogenic differentiation, motor axon growth, and MN specification, with the exact mechanisms underlying these cell interactions remaining to be explored.
–
In conclusion, looking back on how the story started, the loop has been closed by the identification of gene functions that underlie the phenotype which initially attracted my attention. Although my intuition that this had to be connected to the neuronal expression of this atypical Cadherin was not completely wrong after all, conditional mutagenesis nevertheless revealed that this function was modest in regard of the unexpected predominant activity in a cell type – connective tissue – that I had initially not considered. I have learned many lessons along the way of this unusual story. Above all, I learned not to fall in love with a favorite hypothesis but instead to be open to what the facts tell us: when the results of an experiment are the opposite of what you expect, this pushes you to think outside the box, and challenges you to remain unbiased in conceiving experimental plans and analyzing data. It is now time to switch scale, and start exploring what happens at the connective tissue muscle interface from a cell biologist’s point of view.
References
- Caruso, N., et al., Deregulation of the protocadherin gene FAT1 alters muscle shapes: implications for the pathogenesis of facioscapulohumeral dystrophy. PLoS Genet, 2013. 9(6): p. e1003550.
- DeSimone, A.M., et al., Facioscapulohumeral Muscular Dystrophy. Compr Physiol, 2017. 7(4): p. 1229-1279.
- Ciani, L., et al., Mice lacking the giant protocadherin mFAT1 exhibit renal slit junction abnormalities and a partially penetrant cyclopia and anophthalmia phenotype. Mol Cell Biol, 2003. 23(10): p. 3575-82.
- Puppo, F., et al., Identification of variants in the 4q35 gene FAT1 in patients with a facioscapulohumeral dystrophy-like phenotype. Hum Mutat, 2015. 36(4): p. 443-53.
- Mariot, V., et al., Correlation between low FAT1 expression and early affected muscle in facioscapulohumeral muscular dystrophy. Ann Neurol, 2015. 78(3): p. 387-400.
- Helmbacher, F., Tissue-specific activities of the Fat1 cadherin cooperate to control neuromuscular morphogenesis. PLoS Biol, 2018. 16(5): p. e2004734.
- Livet, J., et al., ETS gene Pea3 controls the central position and terminal arborization of specific motor neuron pools. Neuron, 2002. 35(5): p. 877-92.
- Haase, G., et al., GDNF acts through PEA3 to regulate cell body positioning and muscle innervation of specific motor neuron pools. Neuron, 2002. 35(5): p. 893-905.
 (2 votes)
(2 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.