A facial birth defect is caused by perturbation of extreme long-range enhancers
Posted by Hannah Long, on 25 January 2021
By Hannah Long
From the almost identical faces of monozygotic twins, we can appreciate that facial appearance is encoded for the most-part in the DNA of our genomes. Therefore, changes to this DNA sequence can contribute to the variation in facial appearance seen between humans, and at the more extreme end of the spectrum can cause human disease. Consequently, improving our understanding of how genetic sequence encodes for our physical traits remains an exciting and important question in developmental biology.
Interestingly, many of the genetic changes that contribute to facial variation or dysmorphology in disease occur in the non-gene encoding parts of the genome. And this raises the question how non-coding DNA mutations can impact development and drive morphological change. To answer this question, in the Wysocka lab we study facial progenitor cells called the cranial neural crest which give rise to the majority of the facial structures, and investigate how genetic changes can drive alterations in physical traits.
At the beginning of our recent study, published last year in Cell Stem Cell, we set out to determine the molecular mechanisms that cause a human craniofacial disorder, called Pierre Robin sequence (PRS) [1]. In this disorder, under-development of the lower jaw (micrognathia) leads in sequence to posterior displacement of the tongue, and in some cases cleft palate [2, 3] (Figure 1). Several labs had previously utilised genomic methods to identify a number of large deletions and chromosomal translocations in PRS patients that cluster in a gene desert region far upstream of the SOX9 gene .
SOX9 is an important DNA-binding transcription factor that plays crucial roles in the differentiation of numerous cells types, and is expressed broadly during development including in chondrocytes, testis, pancreas, heart valve, lung, kidney, liver, hair follicle stem cells, and progenitors of the face (cranial neural crest) [8]. In keeping with its broad roles during development, heterozygous loss-of-function mutations in the SOX9 gene cause a devastating multisystemic syndrome called campomelic dysplasia. Patients typically exhibit skeletal abnormalities including bowing of the long bones, disorders of sex determination and facial dysmorphism, and occasionally have additional malformations of the heart, lung, kidney and pancreas suggesting differential sensitivity to SOX9 gene dosage (Figure 1). Importantly, selective knockout of Sox9 in mouse facial progenitors results in developmental defects of the facial skeleton [1, 9] emphasizing the importance of Sox9 in the development of the face. Given the non-coding nature of the mutations in PRS patients, it had been suggested that gene regulatory elements called enhancers may be perturbed [4–7]. However, it remained to be functionally demonstrated how these mutations impacted genome function, and whether SOX9 was the target gene to cause this disorder.
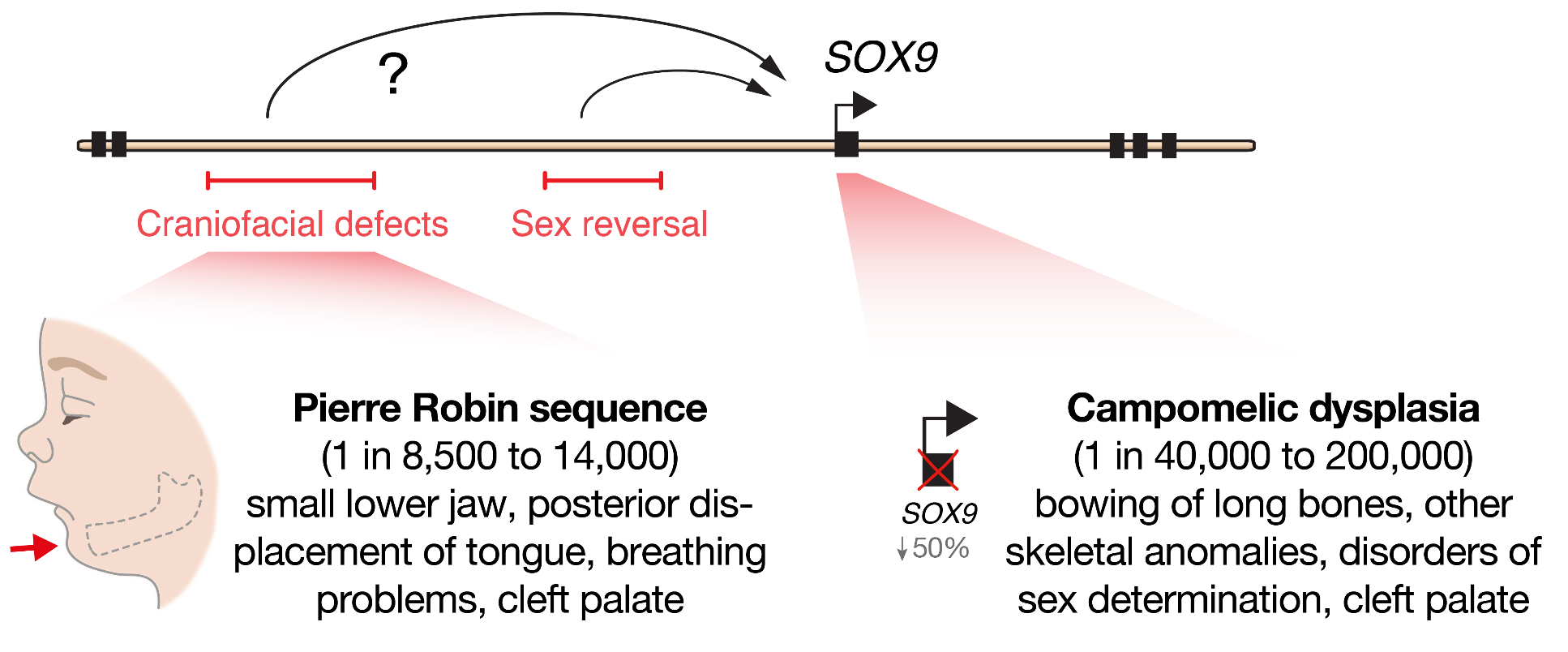
Enhancers are non-coding DNA sequences elements that can act across large genomic distances to regulate gene expression in a cell-type specific manner. And key developmental genes are often associated with complex regulatory landscapes with many such context-dependent enhancers [10]. Therefore, unlike mutations in coding sequences which will alter gene function in all cell-types in which that gene is expressed, mutations in enhancers will have a more developmental stage and cell-type specific impact leading to tissue-restricted phenotypic consequences.
Studying human facial development presents a challenge, as formation of the face occurs early during gestation. However, leveraging a robust in vitro differentiation model of cranial neural crest cells (CNCCs) developed in the lab we have been able to model human craniofacial disorders in culture [11–13]. Utilising this model, and genomic enhancer profiling, we identified three clusters of enhancer elements overlapping the human PRS mutation region. We rationalised that these elements may regulate SOX9 expression during development, and that their loss could cause mis-regulation of SOX9 in the neural crest with detrimental consequences for facial development. Indeed, using CRISPR/Cas9 genome editing we demonstrated that SOX9 expression is specifically perturbated by PRS mutations in the neural crest (and not during cartilage formation when SOX9 is also expressed), defining a developmental window for disease causation in this disorder (Figure 2).
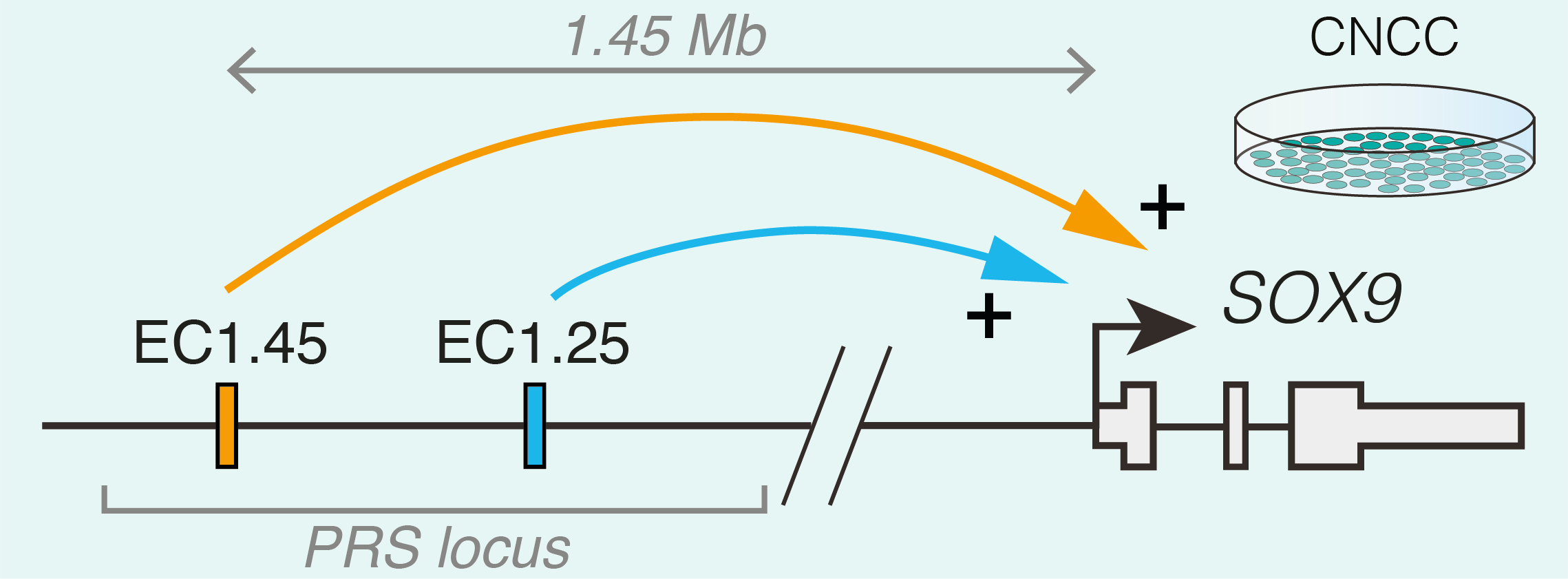
To further understand the morphological impacts of enhancer ablation, we set out to generate mouse models to investigate Pierre Robin sequence disease mechanisms. However, we soon appreciated the challenges associated with modelling human genetic disorders of the non-coding genome in mouse. Unlike genic regions, which make up 1-2% of the genome and are deeply conserved, the remainder of the genome is highly divergent between human and mouse. Indeed across mammalian species, enhancers can vary greatly in their location, sequence composition and activity [14]. Therefore, while one of the PRS-associated enhancer clusters (named EC1.45) was at least partially conserved in activity during mouse facial development, the second enhancer cluster (EC1.25) was not, limiting our further morphological investigation of PRS mutations to EC1.45 alone. Given that directly addressing the functional impact of specific human non-coding mutations in model organisms poses a huge challenge due to a limited sequence conservation of the non-coding genome, future studies exploring the genotype to phenotype connection in human development and disease will greatly benefit from improved models of human development, including advances in organoid models.
Despite the limitations, our models of Sox9 perturbation in mouse led to a number of intriguing observations. Firstly, we determined that lower jaw development is highly sensitized to reduction in Sox9 gene expression compared to the rest of the skull. Therefore, despite the PRS enhancers being widely active across the developing face, this regional sensitivity in the lower jaw to Sox9 gene dosage may drive the phenotypic specificity seen in patients (Figure 3). Secondly, we observed that even small changes in Sox9 expression during development lead to alterations in lower jaw morphology, demonstrating how subtle changes in enhancer activity may drive morphological change and fitness between members of the same species and across evolutionary timescales.

Together, our study uncovers the workings of a complex regulatory domain that controls SOX9 expression during a narrow window of facial development. We further explore the sequence motifs and trans-regulatory factors that regulate enhancer function at the PRS locus, and the conservation of enhancer activity across vertebrate and recent hominin evolution – and we invite you to see our paper for more details. Importantly, the enhancer clusters we characterised at the PRS locus represent the longest-range human enhancers involved in congenital malformations described to date and their deletion in mouse illustrates how small changes in gene expression can lead to morphological variation.
Combined, our work provides a clear illustration and mechanistic details how large deletions and translocations in the non-coding genome can cause human congenital disease. This joins a number of recent studies that have explored how genetic aberrations such as inversions, duplications and topological domain boundary perturbations can either displace enhancers away from their target gene, or can lead to de novo enhancer co-option and ectopic gene expression [15–17].
Looking forward, it is exciting to speculate how such long-range gene regulatory elements (at up to 1.45 megabases away) such as those at the PRS locus find their target gene. From our study, we speculate that perhaps a third element at the PRS locus may play a structural role to bridge these long genomic distances. The developmental importance of this genetic feature, and whether this is a generalisable feature of long-range regulation will be an exciting avenue for future research.
References
- Long HK, Osterwalder M, Welsh IC, et al (2020) Loss of Extreme Long-Range Enhancers in Human Neural Crest Drives a Craniofacial Disorder. Cell Stem Cell 27:765-783.e14. doi: 10.1016/j.stem.2020.09.001
- Robin P (1994) A fall of the base of the tongue considered as a new cause of nasopharyngeal respiratory impairment: Pierre Robin sequence, a translation. 1923. Plast Reconstr Surg 93:1301–1303.
- Tan TY, Kilpatrick N, Farlie PG (2013) Developmental and genetic perspectives on pierre robin sequence. Am J Med Genet Part C Semin Med Genet 163:295–305. doi: 10.1002/ajmg.c.31374
- Benko S, Fantes J a, Amiel J, et al (2009) Highly conserved non-coding elements on either side of SOX9 associated with Pierre Robin sequence. Nat Genet 41:359–364. doi: 10.1038/ng.329
- Amarillo IE, Dipple KM, Quintero-Rivera F (2013) Familial Microdeletion of 17q24.3 Upstream of SOX9 Is Associated With Isolated Pierre Robin Sequence Due to Position Effect. Am J Med Genet Part A 161:1167–1172. doi: 10.1002/ajmg.a.35847
- Gordon CT, Attanasio C, Bhatia S, et al (2014) Identification of Novel Craniofacial Regulatory Domains Located far Upstream of SOX9 and Disrupted in Pierre Robin Sequence. Hum Mutat 35:1011–1020. doi: 10.1002/humu.22606
- Gordon CT, Tan TY, Benko S, et al (2009) Long-range regulation at the SOX9 locus in development and disease. J Med Genet 46:649–56. doi: 10.1136/jmg.2009.068361
- Symon A, Harley V (2017) SOX9: A genomic view of tissue specific expression and action. Int J Biochem Cell Biol 87:18–22. doi: 10.1016/j.biocel.2017.03.005
- Mori-Akiyama Y, Akiyama H, Rowitch DH, de Crombrugghe B (2003) Sox9 is required for determination of the chondrogenic cell lineage in the cranial neural crest. Proc Natl Acad Sci 100:9360–9365. doi: 10.1073/pnas.1631288100
- Long HK, Prescott SL, Wysocka J (2016) Ever-Changing Landscapes: Transcriptional Enhancers in Development and Evolution. Cell 167:1170–1187. doi: 10.1016/j.cell.2016.09.018
- Bajpai R, Chen D a, Rada-Iglesias A, et al (2010) CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463:958–962. doi: 10.1038/nature08733
- Calo E, Gu B, Bowen ME, et al (2018) Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554:112–117. doi: 10.1038/nature25449
- Greenberg RS, Long HK, Swigut T, Wysocka J (2019) Single Amino Acid Change Underlies Distinct Roles of H2A.Z Subtypes in Human Syndrome. Cell 178:1421-1436.e24. doi: 10.1016/j.cell.2019.08.002
- Villar D, Berthelot C, Aldridge S, et al (2015) Enhancer Evolution across 20 Mammalian Species. Cell 160:554–566. doi: 10.1016/j.cell.2015.01.006
- Laugsch M, Bartusel M, Rehimi R, et al (2019) Modeling the Pathological Long-Range Regulatory Effects of Human Structural Variation with Patient-Specific hiPSCs. Cell Stem Cell 24:736-752.e12. doi: 10.1016/j.stem.2019.03.004
- Lupiáñez DG, Kraft K, Heinrich V, et al (2015) Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. doi: 10.1016/j.cell.2015.04.004
- Franke M, Ibrahim DM, Andrey G, et al (2016) Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature 538:265–269. doi: 10.1038/nature19800
 (2 votes)
(2 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.