Computing the worm: artificial intelligence approaches to planarian regeneration and beyond
Posted by Mike Levin, on 30 October 2015
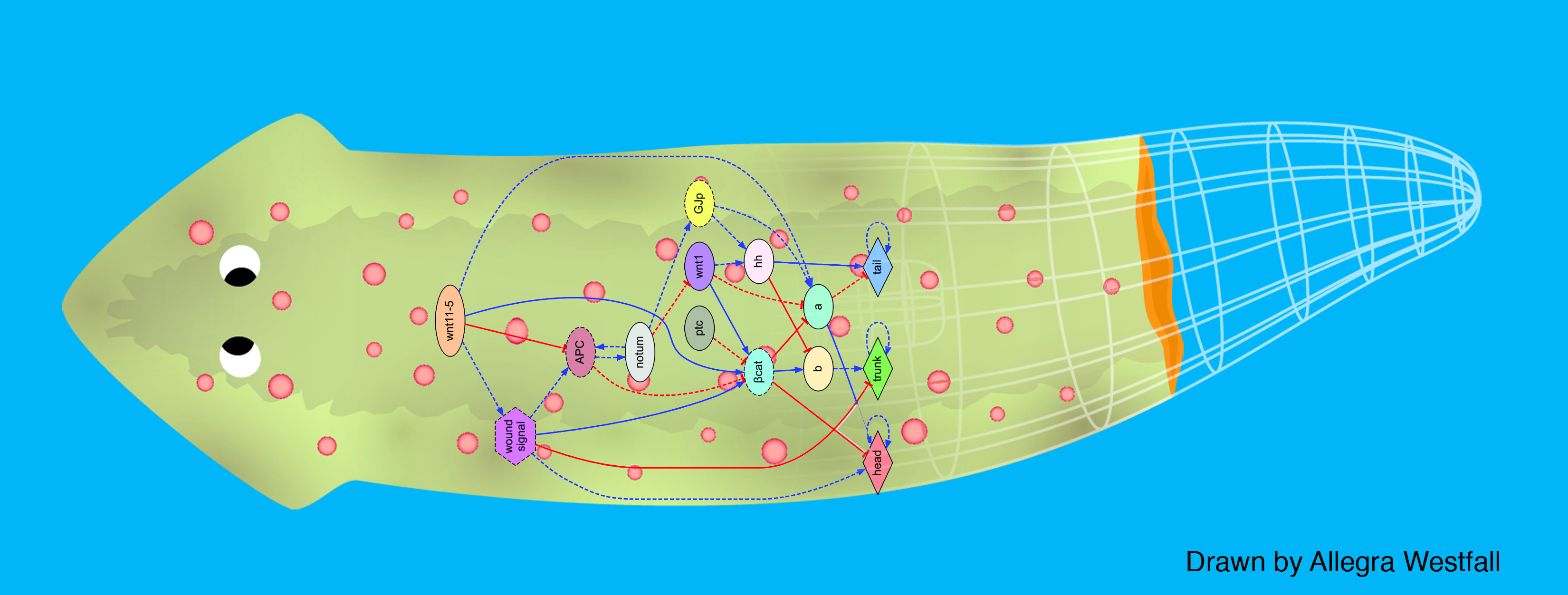
Pattern formation and regulation emerges from cellular activity determined by specific biophysical and genetic rules. A major challenge for developmental biology, biomedicine, and synthetic bioengineering is this highly indirect (Lobo et al., 2014b) relationship between the rules that govern processes at the lower scales, and the anatomical outcomes observed at the macroscopic scale. It is very hard to predict what a complex, iterative, distributed system such as an embryo or a regenerating animal will do, from knowledge of the low-level rules. It is even harder to infer what manipulation can be applied at the molecular level, to achieve a specific anatomical change in biomedical or synthetic morphology applications. This is known as an “inverse problem” in computer science; for example, it’s very easy to iterate a mathematical function like Z<=Z^2+c and create a beautiful fractal (http://mathworld.wolfram.com/MandelbrotSet.html ), but it’s an intractable task to take an arbitrary pattern and figure out what formula would produce it.
In this post, I describe our recent efforts to address the gulf between the molecular details that come out of bench experiments, and the complex patterns or regulatory behavior that we would like to understand and control. We view this as the beginning of a “bioinformatics of shape” – the creation of computational tools addressing shape, not only sequence or expression, and that assist human scientists in discovering models that explain pattern and complex, stochastic developmental phenotypes. An important component of this “robot scientist” (Sparkes et al., 2010) effort is enabling the inference of models that are generative (algorithmic): they specify every step, quantitatively, without ambiguity or magic, and show exactly what dynamics are sufficient to produce the pattern in question. The field is moving towards these kinds of models (Wittmann et al., 2009; Raspopovic et al., 2014; Uzkudun et al., 2015; Werner et al., 2015), and away from simple “arrow diagram” models, that capture valuable loss-of-function and interaction data, but only reveal the pieces necessary for the process to occur. That is generally insufficient to show why or how the complex patterning proceeds to a specific shape, or enable full control of its outcome toward a different goal state; thus, it is crucial to develop tools for generating and testing fully-specified generative models.
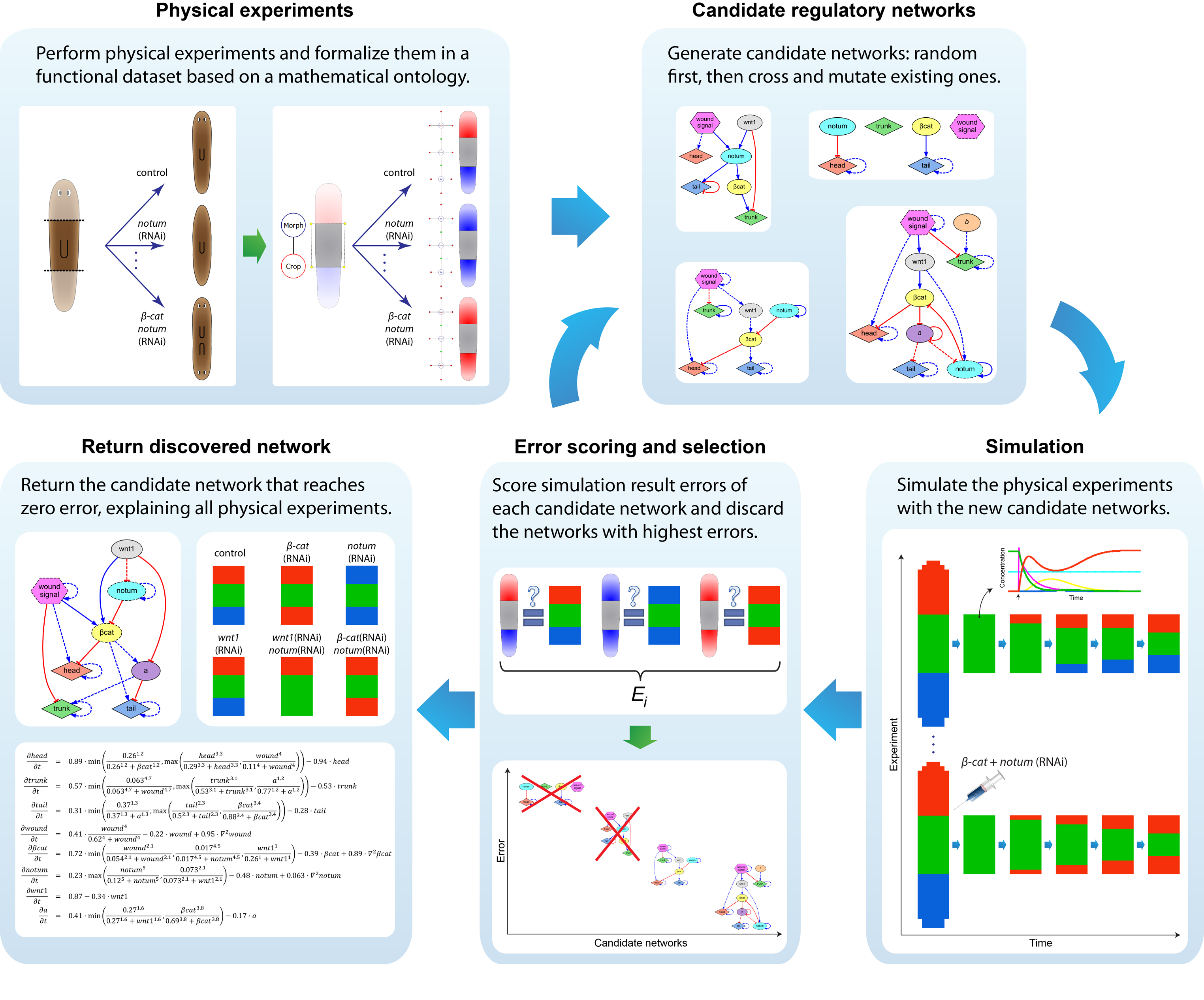
We first tackled the problem of planarian regeneration (Handberg-Thorsager et al., 2008; Lobo et al., 2012). A fragment of these complex bilaterian creatures regenerates all the missing components, no more and no less, in the correct orientation and at the correct locations (Reddien and Sanchez Alvarado, 2004; Gentile et al., 2011). People have been wondering about how this process works for at least 120 years, but despite remarkable molecular advances in stem cell regulation (Reddien et al., 2005; Wagner et al., 2011), there have been no quantitative models that algorithmically explain more than a few different patterning experiments in this very rich body of functional data. We began by creating a formal language (Lobo et al., 2013b) to describe possible experimental manipulations of worms (cuts, grafts, RNAi knockdown, etc.), and a graph representation scheme by which any shape of worm could be encoded (normal, 2-headed, etc.). We then encoded a large number of papers from the planarian literature into a database, which contains the sum total of current knowledge about this model species – a sort of expert system on planarian regeneration, which a human or algorithm can consult when needing to know what outcome results from a given experiment (Lobo et al., 2013a). Next, we created a virtual worm simulator – an in silico platform on which any genetic model of planaria could be run, and manipulated in virtual “experiments”, to see if the predicted outcomes matched the existing results from the database. Models could now be tested, but where to find a model detailed enough to be run on such a simulator? This problem is already so complex that human scientists have a very hard time to simply think of a model whose emergent behavior correctly predicts all the available results. With each new paper published, it becomes harder, not easier, to come up with a model that matches all the data. We thus implemented the final piece: a machine-learning module, that uses evolutionary computation to try to infer a model of gene regulation that behaves just like the real thing, under the published experimental perturbations. The system ran for several days on a supercomputer cluster, and indeed discovered a model that not only explained the existing data but made correct predictions for experiments it had never seen (Lobo and Levin, 2015).
This is the first model of regeneration discovered by an artificial (non-human) intelligence; we and others are testing the model’s predictions and its ability to suggest manipulations that result in never-before-seen phenotypes (as well as extending the platform to limb regeneration in multiple species (Lobo et al., 2014a)). Remarkably, the model it found was not an incomprehensible hairball (akin to diagrams of “metabolism”) but a small, easy-to-understand network with only two unknowns (which have since been identified from their connections to neighbors in protein interactome databases). The power of this system is that any future papers’ data can be continuously be added to the database, and the inference process re-run, to discover ever better models. Regardless of the fate of this particular model, we think this platform is a proof-of-concept, highly generalizable system for using artificial intelligence to assist scientists in the most creative aspect of our work: trying to come up with a testable model of what could be going on during complex pattern regulation.
References
Gentile, L., Cebria, F. and Bartscherer, K. (2011) ‘The planarian flatworm: an in vivo model for stem cell biology and nervous system regeneration’, Dis Model Mech 4(1): 12-9,
http://dmm.biologists.org/content/4/1/12.full.pdf
Handberg-Thorsager, M., Fernandez, E. and Salo, E. (2008) ‘Stem cells and regeneration in planarians’, Front Biosci 13: 6374-94, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=18508666
Lobo, D., Beane, W. S. and Levin, M. (2012) ‘Modeling planarian regeneration: a primer for reverse-engineering the worm’, PLoS Comput Biol 8(4): e1002481, http://www.ncbi.nlm.nih.gov/pubmed/22570595
Lobo, D., Feldman, E. B., Shah, M., Malone, T. J. and Levin, M. (2014a) ‘A bioinformatics expert system linking functional data to anatomical outcomes in limb regeneration’, Regeneration: n/a-n/a, http://dx.doi.org/10.1002/reg2.13
Lobo, D. and Levin, M. (2015) ‘Inferring Regulatory Networks from Experimental Morphological Phenotypes: A Computational Method Reverse-Engineers Planarian Regeneration’, PLoS Comput Biol 11(6): e1004295, http://www.ncbi.nlm.nih.gov/pubmed/26042810
Lobo, D., Malone, T. J. and Levin, M. (2013a) ‘Planform: an application and database of graph-encoded planarian regenerative experiments’, Bioinformatics http://www.ncbi.nlm.nih.gov/pubmed/23426257
Lobo, D., Malone, T. J. and Levin, M. (2013b) ‘Towards a bioinformatics of patterning: a computational approach to understanding regulative morphogenesis’, Biology Open 2(2): 156-69, http://www.ncbi.nlm.nih.gov/pubmed/2342966
Lobo, D., Solano, M., Bubenik, G. A. and Levin, M. (2014b) ‘A linear-encoding model explains the variability of the target morphology in regeneration’, Journal of the Royal Society, Interface / the Royal Society 11(92): 20130918, http://www.ncbi.nlm.nih.gov/pubmed/24402915
Raspopovic, J., Marcon, L., Russo, L. and Sharpe, J. (2014) ‘Modeling digits. Digit patterning is controlled by a Bmp-Sox9-Wnt Turing network modulated by morphogen gradients’, Science 345(6196): 566-70, http://www.ncbi.nlm.nih.gov/pubmed/25082703
http://www.sciencemag.org/content/345/6196/566.full.pdf
Reddien, P. W., Oviedo, N. J., Jennings, J. R., Jenkin, J. C. and Sanchez Alvarado, A. (2005) ‘SMEDWI-2 is a PIWI-like protein that regulates planarian stem cells’, Science 310(5752): 1327-30,
Reddien, P. W. and Sanchez Alvarado, A. (2004) ‘Fundamentals of planarian regeneration’, Annu Rev Cell Dev Biol 20: 725-57, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=15473858
Sparkes, A., Aubrey, W., Byrne, E., Clare, A., Khan, M. N., Liakata, M., Markham, M., Rowland, J., Soldatova, L. N., Whelan, K. E. et al. (2010) ‘Towards Robot Scientists for autonomous scientific discovery’, Autom Exp 2: 1, http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=20119518
Uzkudun, M., Marcon, L. and Sharpe, J. (2015) ‘Data-driven modelling of a gene regulatory network for cell fate decisions in the growing limb bud’, Molecular Systems Biology 11(7): 815, http://www.ncbi.nlm.nih.gov/pubmed/26174932
Wagner, D. E., Wang, I. E. and Reddien, P. W. (2011) ‘Clonogenic neoblasts are pluripotent adult stem cells that underlie planarian regeneration’, Science 332(6031): 811-6, http://www.sciencemag.org/content/332/6031/811.full.pdf
Werner, S., St√ºckemann, T., Beir√°n Amigo, M., Rink, J. C., J√ºlicher, F. and Friedrich, B. M. (2015) ‘Scaling and Regeneration of Self-Organized Patterns’, Physical Review Letters 114(13): 138101, http://link.aps.org/doi/10.1103/PhysRevLett.114.138101
Wittmann, D. M., Blochl, F., Trumbach, D., Wurst, W., Prakash, N. and Theis, F. J. (2009) ‘Spatial analysis of expression patterns predicts genetic interactions at the mid-hindbrain boundary’, PLoS Comput Biol 5(11): e1000569, http://www.ncbi.nlm.nih.gov/pubmed/19936059
 (2 votes)
(2 votes)6 thoughts on “Computing the worm: artificial intelligence approaches to planarian regeneration and beyond”
Leave a Reply
Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.
Dear Dr. Levin,
Since you published this paper, how many interesting NEW discoveries have you made? I’m afraid ‘inverse problems’ are not solvable unless you feed them new a priori knowledge. Moreover, your approach tries to bridge phenotypic morphologies with gene networks skipping the cell level which is considered homogeneous. To me, this is rather odd.
Dear Dr. Baguñà,
I see several different questions in your comment; let me try to address them:
> Since you published this paper, how many interesting NEW discoveries have you made?
since the publication of that paper, we have published about 16 new papers, and I hope at least some of them describe interesting new discoveries. But perhaps you are asking, how many new papers has this particular paper made possible. This is a good question, although perhaps it’s better to ask that question in a few years’ time: the system we describe, and the model it uncovered, have only existed since 2015 – less than a year. Surely not enough time to gauge its impact, if that is the point of the question. However, I am happy to tell you that the model makes a few unique, novel predictions which we have recently validated in planaria at the bench, and are writing the manuscript up now. We appreciate your enthusiasm in seeing the results come out and will publish it as soon as humanly possible. I’m sure you understand that testing models in planaria with good functional data can take a year; I believe we are not being unreasonably slow. We encourage others in the planarian community to do so as well – our system facilitates other labs to mine the data for predictive models and test them.
I can also tell you that our other recent paper, showing a similar machine learning platform’s inference of melanoma dynamics in vertebrates (http://stke.sciencemag.org/cgi/content/full/sigtrans;8/397/ra99?ijkey=DNnSge7tn.tcE&keytype=ref&siteid=sigtrans ) also generated predictions that enabled us to produce a desired phenotype never before described; the AI-discovered model actually enabled a new capability and suggested the exact reagents that can be used to implement that desired outcome in Xenopus tadpoles. Our manuscript describing this is likewise in preparation and we look forward to sharing it with the community.
In any case, we are in agreement that it is important to test models empirically, and we’re doing just that. I also welcome any practical advice on how to make the resulting papers come out more rapidly.
> I’m afraid ‘inverse problems’ are not solvable unless you feed them new a priori knowledge
well, we agree on the difficulty of inverse problems ( http://rsif.royalsocietypublishing.org/cgi/reprint/rsif.2013.0918?ijkey=r6H7rxetYCz9r9k&keytype=ref ), however it is not obvious to me what you mean by “new a priori knowledge”. Human scientists work on the assumption that by looking at existing data, they can infer a predictive model of what’s going on. I think it’s premature to assert that this process has some ineffable quality that is unreachable by machine learning and requires humans at every step. It may turn out to be the case, but it’s way too soon to assert that without trying to provide tools to augment human scientists’ efforts. Too many other problems have been thought of as human-only, and then shown to benefit from computerized tools. What scientists do is look at data, and try to come up with a model that explains that data. That is what our machine learning platform does. Testing those models is then done by humans at the bench, but given the dearth of comprehensive models in this field, I would think a computational tool to provide candidate models for us to test would be a welcome addition to the toolkit.
> Moreover, your approach tries to bridge phenotypic morphologies with gene networks
I certainly do not claim that gene networks are sufficient to explain morphogenesis. Indeed many of our papers address additional biophysical systems that interact with gene networks to regulate patterning (http://ase.tufts.edu/biology/labs/levin/publications/bioelectricity.htm ). However, one has to start somewhere – if one tries to model every detail simultaneously, no progress will be made. Since many people are indeed interested in inferring gene regulatory networks for pattern formation (as evidenced by numerous papers in which the last figure is some sort of GRN model), we produced a tool that facilitates this process. We started with gene networks, but I am in complete agreement with you that this is not the end, and we are in the process of augmenting our simulator to include several there signaling modalities. This is a significant undertaking, and requires a lot of work. But now that the GRN component works, we can turn our attention to the next step which will broaden the framework beyond gene networks.
> skipping the cell level which is considered homogeneous
you are absolutely correct in that our model did not model individual cells (just as many gradient models in developmental biology do not). We did this as a simplifying first step, and future versions of this system will explicitly represent cells as part of including bioelectric signaling. However, I point out that it is a reasonable strategy to see how far one can get with a minimum of complexity in a model. It remains to be seen whether a spatialized model does better than ours in explaining the published results on planarian regeneration; for now, our system seems to have found a fairly high-quality model (not claiming “the correct” model, as no model is), without using discretization of cells. We look forward to comparing it with any alternatives you or other labs may produce with different methods.
I hope that addresses your thoughtful questions; thank you for your interest in our work,
Mike Levin
Here’s one of our recent in vivo tests of the novel predictions of the computational model:
http://bioinformatics.oxfordjournals.org/content/early/2016/05/09/bioinformatics.btw299.abstract?keytype=ref&ijkey=JunJoareh3qrVOZ
A few others are in the pipeline.
Thanks. A comment will follow soon.
Jaume Baguñà
Sorry for the delay. I read it through but I could not grasp it entirely. Three comments/questions. First, I do not understand hnf4- worms having a WT phenotype. If hnf4+ gene is a key gene for gut, RNAi against it should heavily distort the central body region which it is not. Why is it so? Second, I don’t see much difference between tail areas between hnf4- and hh- worms except that the first has a larger blastema than the second that likely giving the statistical significance in Fig 3. Besides hh- animals are axially shifted (distorted). Third, and most importantly, I would like to see the same set of experiments using heads and tails instead of trunk regions. Are you planning to do them?
Many thanks.
Dear Dr. Levin,
On May 26th 2016 I posted a comment on your new paper in Bioinformatics 2016 with, so far, no answer. Whenever you feel like, I would appreciate a short (or long) answer. Thanks a lot.
Jaume Baguñà