Functional interrogation of the C-terminal domain of RNA polymerase II in fruit flies
Posted by feiyue.lu92, on 24 April 2019
Feiyue Lu and David Gilmour tell the story behind their recent paper in Molecular Cell
RNA polymerase II (Pol II) is the enzyme responsible for transcribing most genes in eukaryotes. The C-terminal domain (CTD) is a highly repetitive, unstructured domain on the largest Pol II subunit, Rpb1. It consists of numerous repeats of seven amino acids and serves as a binding platform for numerous factors involved in gene regulation, including chromatin modifying enzymes, transcription factors and RNA processing enzymes. Both length and sequence complexity of the CTD greatly vary among species of different lineages. For example, the CTD of S. cerevisiae has 26 repeats, the majority of which are consensus repeats of YSPTSPS. In contrast, mammalian CTDs have 52 repeats, the distal repeats are mostly divergent repeats whose sequence differ from the consensus at one or more positions.
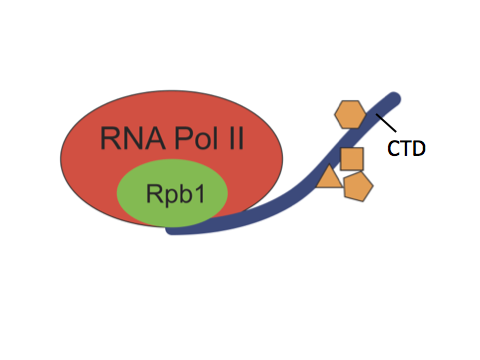
The length and sequence complexity of the CTD were thought to regulate gene expression and be essential for the development of complex organisms. In the past decade, this proposition has been reinforced by several studies in mammalian cells. These studies showed that post-translational modifications occur on certain residues of the divergent repeats, some of which are recognized by factors only present in higher eukaryotes, and that mutations which prevent such modifications lead to misexpression of genes. Yet the significance of the divergent residues has never been systematically examined in the context of development.
Considering the above findings in mammalian cells, one would anticipate that a mouse would die if all of its CTD repeats were replaced with the consensus repeats that predominate the yeast CTD. However, generating transgenic mouse lines is very time and cost consuming. Instead, we decided to perform systematic mutagenesis of the CTD in Drosophila, an organism that is also highly genetically malleable but faster and cheaper to mutate compared to mice. In addition, 40 of the 42 CTD repeats in the fly diverge from the consensus, so it is an ideal model to interrogate the significance of divergent repeats.
The project was started at a time when CRISPR/Cas9 was not yet widely adopted by the fly research community, so initially we relied on RNAi knockdown to deplete the endogenous Rpb1 subunit. We also tested if the co-expression of an RNAi-resistant Rpb1 harboring mutations in the CTD would rescue the RNAi phenotype, making approximately 20 transgenic fly lines expressing various mutant forms of Rpb1 in an attempt to identify essential regions of the fly CTD. To our surprise, despite the high conservation in the amino acid sequence of the CTD within the fruit fly genus, most of our mutant CTD flies were viable, even when internal deletions removed up to 30% of the entire CTD. The only essential region we identified was an 8-repeat region encompassing the only two consensus repeats in the fly CTD (Gibbs et al., 2017). This suggests that the majority of the divergent repeats in the fly CTD are redundant.
Our initial systematic mutagenesis of the fly CTD emboldened us to test the idea that all divergent repeats could be replaced with consensus repeats. However, most previous findings in mammalian cells argue that the divergent CTD repeats are essential, so we still expected that flies would die with an all-consensus CTD. The most straightforward way to test this would be to simply replace the fly CTD with 42 consensus repeats. However, since many of the CTD repeats appear redundant, consensus repeats might be able to do what divergent repeats do, but with more or fewer repeats to achieve the same function. Therefore, we tested a series of consensus CTDs ranging from 10 to 52 repeats.
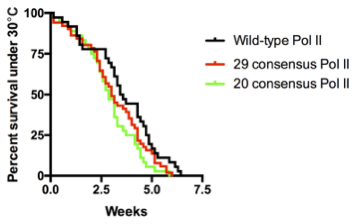
Surprisingly, we obtained normal looking flies with solely consensus repeats. This was achieved with only 20 to 29 repeats, which approximates the length of the yeast CTD. In contrast, flies with 42 consensus repeats, which matches the length of the wild-type fly CTD, barely survived to adulthood. In hindsight, we were fortunate to have tested varying numbers of consensus repeats because had we only tested the 42 consensus repeat CTD, we would have concluded that the divergent motifs are indispensable, which would have driven the project in a different direction. Additionally, flies with 52 consensus repeats died, whereas flies with the human CTD, which is also composed of 52 repeats but contains a mixture of consensus and divergent repeats, were able to survive. Therefore, it seems as though consensus repeats alone are sufficient, yet having too many of them is bad for fly development.
We were shocked by the results of the RNAi rescue experiment but at the same time concerned that residual endogenous Rpb1 might contribute to survival. Therefore, we decided to use CRISPR/Cas9 to mutate the endogenous Rpb1 gene. Remarkably, the CRISPR results agreed with our RNAi experiment. In particular, our flies with 20 to 29 consensus repeats did just as well as flies with the wild-type CTD even under temperature stress.
As flies can survive with solely consensus repeats, consensus and divergent repeats should both be able to interact with the same group of factors. Our data also suggests that interactions provided by the consensus repeats are likely stronger, since in the case of an all-consensus CTD, fewer than half the number of wild-type repeats are needed for such interactions to occur properly. This fits nicely with the phase separation mechanism proposed by Weber and Brangwynne where protein interactions could be mediated by weak, multivalent interactions (Weber and Brangwynne, 2012). In reviews published when our project was being developed, Hnisz et al and Harlen et al proposed that such forces could also drive CTD:factor interactions (Harlen and Churchman, 2017; Hnisz et al., 2017). Also consistent with the phase separation theory was that the deleterious effects of having too many consensus repeats could be reverted either by shortening the CTD (as was the case with our truncated consensus CTDs), or by replacing some of the stronger repeats with weaker ones (as was the case with 52 consensus CTD versus the human CTD), both of which could reduce the overall valency of the CTD.
Coincidentally, our co-author Bede Portz had a chat with Stirling Churchman at the Cold Spring Harbor Mechanisms of Eukaryotic Transcription Meeting about our CTD mutagenesis in flies. Stirling was interested in our findings and was curious to know if the CTD by itself could target transcription sites. Bede thought that the fruit fly salivary glands would be an ideal system to test this. The salivary gland cells undergo rounds of replication without cell division, giving rise to polytene chromosomes where numerous copies of sister chromatids are fused together, which allows for visualization of each transcription site as a distinct band (or ‘puff’). Before us, John Lis’ group had shown that many transcription components such as Pol II are compartmentalized at heat shock puffs in salivary glands (Yao et al., 2006; Yao et al., 2007; Zobeck et al., 2010). Transcription compartments have also been documented recently using live super-resolution microscopy or in other systems where numerous copies of the same DNA sequences were introduced into a genomic location (Cho et al., 2018; Chong et al., 2018). However, the polytene chromosomes provide the power to visually determine a factor’s spatial location with respect to euchromatin, heterochromatin and nucleoplasm without having to rely on super-resolution microscopy or amplifying the number of copies of a candidate gene.
We fused just the CTD to GFP and expressed it in salivary glands. Interestingly, GFP-CTD colocalizes with Pol II on fixed polytene chromosome spreads. One possibility is that the CTD alone dynamically associates with the transcription sites. The alternative is that the GFP-CTD is stably bound by CTD binding partners that are abundant at transcription sites. To distinguish the two possibilities, we turned to live imaging and showed that GFP-CTD on puffs can rapidly recover upon photobleaching, suggesting that the CTD is dynamically recruited to transcription compartments. In addition, phase separation is likely the driving force for the behavior of the CTD, as 1,6-hexanediol, which has been shown to disrupt weak hydrophobic interactions, can disrupt the association of GFP-CTD to chromosomes.
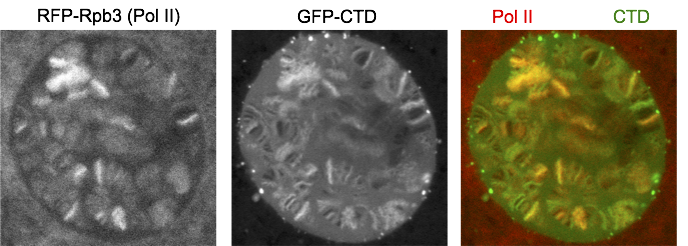
The partitioning characteristic of a molecule is influenced by its valency. This led us to investigate how the number of consensus repeats impacts the behavior of the CTD. To address this, we fused our series of consensus CTDs to GFP. Phase separation models predict that changes in valency could lead to pathological aggregates (Weber and Brangwynne, 2012). Indeed, our 42 and 52 repeats of consensus CTDs, which do not support normal function in the fly, showed less GFP on puffs and correspondingly more extrachromosomal foci when fused to GFP. Much of the extrachromosomal foci were neither recovered upon photobleaching nor dispersed by 1,6-hexanediol, suggesting that they were stable aggregates. Interestingly, none of our consensus CTDs partition into transcription compartments as readily as the wild-type CTD. These results suggest that the divergent repeats play a role in either preventing aggregation, or facilitating partitioning into transcription compartments, both of which could be forces that constrain the sequence conservation of the CTD in evolution.
At the start of this project, we had anticipated that the divergent repeats would be essential for fly viability since the sequences of the CTD in 12 species of Drosophila are highly conserved. Moreover, the ratio of synonymous to nonsynonymous mutations among the 12 species is high, thus indicating that the fly CTD is under significant purifying selection. Our discovery that the entire fly CTD can be replaced by consensus repeats poses a conundrum since it calls into question the basis for the purifying selection. One possibility is that intra and intermolecular interactions intrinsic to the CTD constrain the sequence. Our SAXs analysis showed that the CTD adopts a compact random coil structure and the compaction implies transient interactions within the CTD (Gibbs et al., 2017). Also, recent results show that CTD molecules self-associate to form droplets (Boehning et al., 2018). Both types of interactions might need to be finely tuned to prevent aggregation and allow for factor binding to the CTD. Despite the sequence differences, the 29 consensus and wild-type Drosophila CTDs appear to meet these constraints.
The theory of constructive neutral evolution provides another answer to the conundrum. This theory was formulated to explain the gap between the simplicity and complexity of molecular machines that serve the same function in different organisms (Gray et al., 2010). In our case, the 29 consensus CTD represents the simple end of the spectrum while the Drosophila CTD represents the complex end. For both to be functionally equivalent, we posit that the essential functions of the CTD are mediated by a limited number of proteins that interact with the consensus repeats. It is notable that while the Drosophila CTD only contains two repeats that exactly match the consensus, these are embedded in a region encompassing seven other near-consensus motifs. Deletion of this region completely eliminated the capacity of Rpb1 to support fly viability. Constructive neutral evolution posits that chance mutations in the consensus sequences are tolerated by the fortuitous binding of other proteins which compensate for the loss of the consensus sequences. This fortuitous interaction might offset the tendency of the mutation to cause the CTD to aggregate or become mistargeted. Alternatively, the loss of consensus repeat could diminish the affinity of an essential protein for the CTD but this could be offset by the fortuitous binding of another protein that simultaneously associates with the mutant repeat and the protein that normally binds directly to the consensus repeat. Occurrence of the fortuitous binding caused by one mutation sets the stage for mutating additional consensus repeats. The evolutionary trajectory is then dictated by chance and could explain why the consensus CTD, the human CTD, and the Drosophila CTD all support Rpb1 function in the fly. If the consensus CTD truly represents the simplest CTD, then there should exist a class of mutations in genes outside of the Rpb1 gene that are deleterious to wild-type flies but not to those carrying the 29 consensus CTD.
Considering the number of CTD mutants that we had to generate to come to our final conclusion, we feel extremely lucky to have started this project in flies: the fruit fly is indeed an amazing model system to study the CTD. In addition to this work, the divergent nature of the fly CTD has allowed our collaborators Dr. Scott Showalter at PSU, Drs. Yan “Jessie” Zhang and Jennifer Brodbelt to map structural changes and post-translational modifications to each individual repeat without having to introduce additional mutations to the CTD (Gibbs et al., 2017; Mayfield et al., 2016). Furthermore, the fly salivary gland present a unique system to characterize the spatial distribution of transcription factors with just a standard confocal microscope. We envision that the currently available gene-editing and optogenetic tools will allow more exciting discoveries of transcription to be made in the salivary glands.
References
Boehning, M., Dugast-Darzacq, C., Rankovic, M., Hansen, A. S., Yu, T., Marie-Nelly, H., McSwiggen, D. T., Kokic, G., Dailey, G. M., Cramer, P., et al. (2018). RNA polymerase II clustering through carboxy-terminal domain phase separation. Nat. Struct. Mol. Biol.
Cho, W.-K., Spille, J.-H., Hecht, M., Lee, C., Li, C., Grube, V. and Cisse, I. I. (2018). Mediator and RNA polymerase II clusters associate in transcription-dependent condensates. Science.
Chong, S., Dugast-Darzacq, C., Liu, Z., Dong, P., Dailey, G. M., Cattoglio, C., Heckert, A., Banala, S., Lavis, L., Darzacq, X., et al. (2018). Imaging dynamic and selective low-complexity domain interactions that control gene transcription. Science 361,.
Gibbs, E. B., Lu, F., Portz, B., Fisher, M. J., Medellin, B. P., Laremore, T. N., Zhang, Y. J., Gilmour, D. S. and Showalter, S. A. (2017). Phosphorylation induces sequence-specific conformational switches in the RNA polymerase II C-terminal domain. Nat. Commun. 8, 15233.
Gray, M. W., Lukes, J., Archibald, J. M., Keeling, P. J. and Doolittle, W. F. (2010). Irremediable Complexity? Science 330, 920–921.
Harlen, K. M. and Churchman, L. S. (2017). The code and beyond: transcription regulation by the RNA polymerase II carboxy-terminal domain. Nat. Rev. Mol. Cell Biol. 18, 263–273.
Hnisz, D., Shrinivas, K., Young, R. A., Chakraborty, A. K. and Sharp, P. A. (2017). A Phase Separation Model for Transcriptional Control. Cell 169, 13–23.
Mayfield, J. E., Robinson, M. R., Cotham, V. C., Irani, S., Matthews, W. L., Ram, A., Gilmour, D. S., Cannon, J. R., Zhang, Y. J. and Brodbelt, J. S. (2016). Mapping the phosphorylation pattern of drosophila melanogaster RNA polymerase ii carboxyl-terminal domain using ultraviolet photodissociation mass spectrometry. ACS Chem. Biol. 12, 153–162.
Weber, S. C. and Brangwynne, C. P. (2012). Getting RNA and protein in phase. Cell 149, 1188–1191.
Yao, J., Munson, K. M., Webb, W. W. and Lis, J. T. (2006). Dynamics of heat shock factor association with native gene loci in living cells. Nature 442, 1050–1053.
Yao, J., Ardehali, M. B., Fecko, C. J., Webb, W. W. and Lis, J. T. (2007). Intranuclear distribution and local dynamics of RNA polymerase II during transcription activation. Mol. Cell 28, 978–990.
Zobeck, K. L., Buckley, M. S., Zipfel, W. R. and Lis, J. T. (2010). Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol. Cell 40, 965–975.
 (2 votes)
(2 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.