Enabling research with human embryonic and fetal tissue resources
Posted by the Node, on 24 September 2015
This Spotlight article was written by Dianne Gerrelli, Steven Lisgo, Andrew J. Copp and Susan Lindsay, and was first published in Development.
Congenital anomalies are a significant burden on human health. Understanding the developmental origins of such anomalies is key to developing potential therapies. The Human Developmental Biology Resource (HDBR), based in London and Newcastle, UK, was established to provide embryonic and fetal material for a variety of human studies ranging from single gene expression analysis to large-scale genomic/transcriptomic studies. Increasingly, HDBR material is enabling the derivation of stem cell lines and contributing towards developments in tissue engineering. Use of the HDBR and other fetal tissue resources discussed here will contribute to the long-term aims of understanding the causation and pathogenesis of congenital anomalies, and developing new methods for their treatment and prevention.
Introduction
An important goal of developmental biology is to understand human embryonic/fetal development and the causes of congenital anomalies. A better understanding of the gene pathways that lead to developmental anomalies will aid new medical approaches for disease treatment and prevention. The causes of congenital anomalies include a variety of genetic and environmental factors, with the majority probably involving multifactorial aetiology. In Europe the recorded prevalence of major congenital anomalies is 23.9 per 1000 pregnancies, of which 80% were live births and 20% resulted in termination, fetal death or stillbirth (Dolk et al., 2010). Children who survive with congenital anomalies frequently experience long-term disability. For example, patients with congenital heart defects (the commonest group of anomalies) may develop severe disability in the first few days after birth (e.g. transposition of the great vessels), often requiring early surgery. In economic terms, the lifetime cost of caring for someone with a severe birth defect, like spina bifida, is estimated at over $0.5 million (Yi et al., 2011). This represents a challenge not only for individuals and families, but also for healthcare systems. In recent decades it has become possible to prevent some congenital anomalies. Two notable examples are vaccination of women against rubella to reduce the number of newborn babies with congenital rubella syndrome (Tookey and Peckham, 1999), and supplementation with folic acid in early pregnancy to decrease the prevalence of neural tube defects (Eichholzer et al., 2006).
Animal models are widely used to test hypotheses about the development of the embryo and fetus. Over the last 10 years there have been huge advances in the understanding of model organisms, in terms of whole-genome sequencing, identifying gene regulatory networks and determining developmental mechanisms. A striking finding is that the majority of protein-coding genes are shared between mouse and human (Yue et al., 2014). Therefore, the differences between the species are unlikely to be due to gene diversity but mainly to modifications in regulatory programmes controlling where and when genes are expressed. The spatial and temporal control of gene expression is therefore extremely important and might help to explain what makes us human.
As well as similarities, there are many established differences between the development of humans and model organisms such as the mouse. Most notably, during evolution the brain has changed in size and shape. Differences are particularly evident in the development of the human cerebral cortex (Bae et al., 2015), with huge increases in the number of cells and in the complexity of cell types. The cortex of the human brain also undergoes a process of folding known as gyrification, which increases the number of cortical neurons. This gyrification is thought to correlate with increased cognitive abilities (Gautam et al., 2015). The mouse brain does not undergo gyrification.
As another example, the eyes of humans are also more complex: they contain three types of cone cells as opposed to only two in mice; while the macula – a highly pigmented area at the back of the human retina – is absent in mice. A number of genes whose mutation is associated with human congenital syndromes, such as KAL1 (ANOS1) (Cadman et al., 2007) and SHOX (Blaschke and Rappold, 2006), are not found in the mouse genome. Thus, although model organisms have proved hugely valuable in understanding developmental processes, there is also a need to study human tissue directly.
Here, we present an overview of the resources available to the research community for analysing human development, with a particular focus on the Human Developmental Biology Resource (HDBR). Resources such as tissue banks, bioinformatics portals and archive collections provide researchers with crucial information and material for the analysis of human development and associated congenital abnormalities.
Research tissue banks
Several research tissue banks have been created in the UK and elsewhere to enable researchers to gain access to human embryonic and fetal samples. These banks collect tissue in a systematic fashion and conform to the highest ethical standards and research governance.
The Human Developmental Biology Resource (HDBR)
The HDBR (www.hdbr.org; with whom the authors are associated) was established in 1999 and is funded by the Medical Research Council and Wellcome Trust. It has National Research Ethics Service approval and is licensed by the UK Human Tissue Authority (https://www.hta.gov.uk/). The collection of material from elective terminations of pregnancy currently comprises over 4000 specimens aged between 3 and 20 post-conception weeks (pcw). The resource continues to collect material, with 400 new specimens added each year. All specimens are karyotyped, with 4% having chromosomal abnormalities (most commonly trisomy 21 and monosomy X) and 9% displaying some form of phenotypic abnormality.
Various types of sample are available from the HDBR (Fig. 1). The majority of tissue provided by the resource is from chromosomally normal samples but material can also be provided from specimens with chromosomal abnormalities. Fresh tissue from specific organs and developmental ages can be used in a range of scientific procedures, such as fluorescence-activated cell sorting of live cells to purify specific human fetal cell populations in order to derive primary cell lines or stem cells. Frozen tissue can be used for the production of mRNA, genomic DNA or protein. These samples are being used in large-scale transcriptome analyses to compare human and animal models and across specific tissues, sexes and between normal and karyotypically abnormal samples.
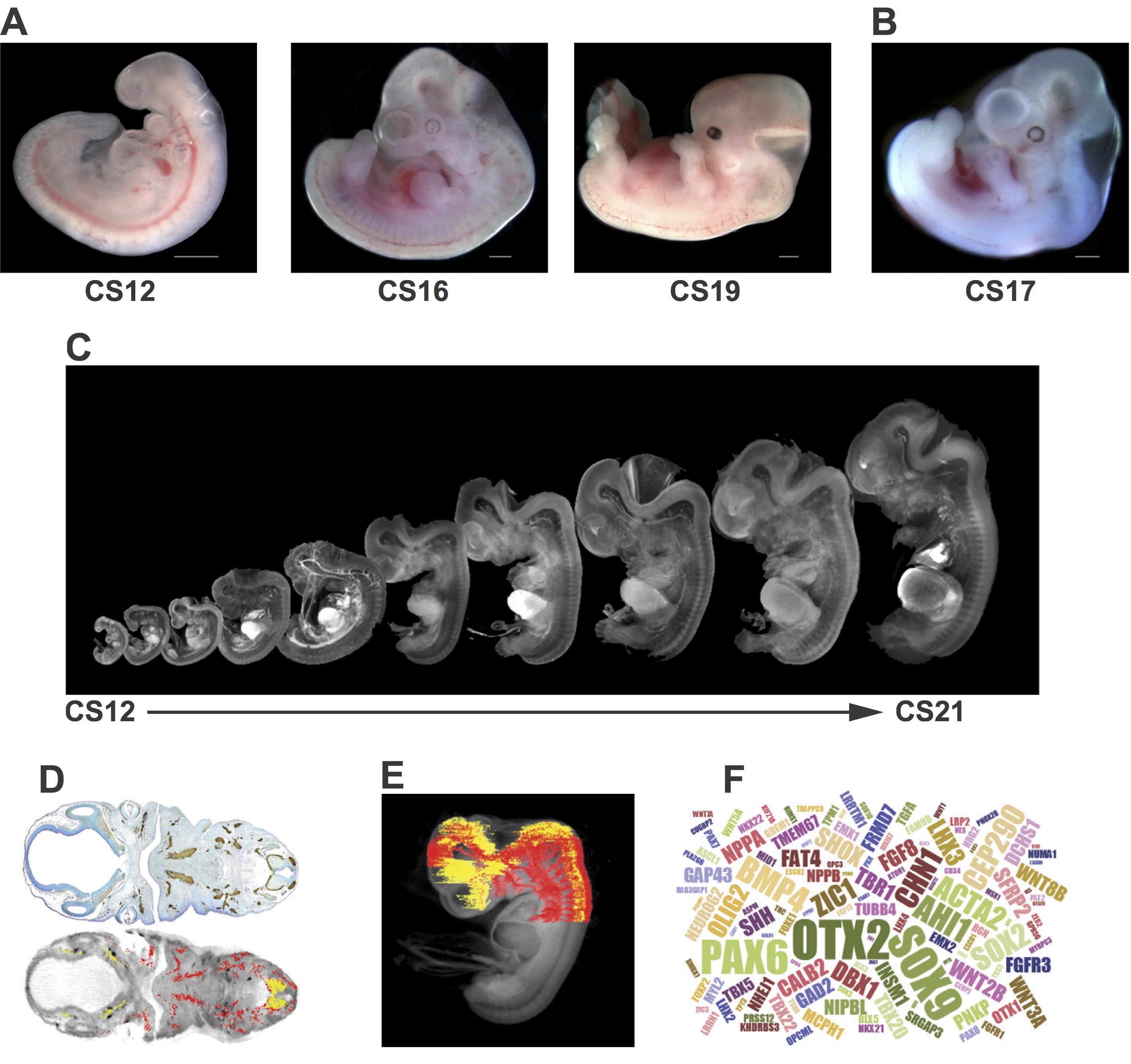
Fig. 1 Examples of resources available from the HDBR. (A) Images of embryos at (from left to right) CS12 [∼26 days post conception (dpc)], CS16 (∼37 dpc) and CS19 (∼47 dpc). (B) A CS17 (∼41 dpc) embryo with trisomy 21. (C) CS12-CS21 series of 3D models, generated using optical projection tomography (OPT) from embryos in the HDBR collection, displayed at their relative sizes. Movies and images of the models can be viewed or the full models requested via the HuDSeN website (www.hudsen.org). (D) The HDBR provides access to curated gene expression data. The upper image shows a section through a CS17 embryo stained with anti-GAP43 antibody (brown); the lower image is of the corresponding section in the CS17 OPT model onto which the GAP43 expression data have been mapped (red, strong expression; yellow, moderate expression). Expression data were mapped from experimental sections to digital sections using MAPaint software (http://www.emouseatlas.org/emap/home.html). (E) 3D expression domain of GAP43 expression in the head and part of the body built up by mapping data from sections as shown in D. Experimental and mapped data are uploaded to a spatial database (www.hudsen.org). (F) Word cloud representation of the expression data available in the HuDSeN human gene expression spatial database (www.hudsen.org). The word cloud represents the number of entries for each gene and there are currently data from 128 genes. Scale bars: 1 mm.
The HDBR also provides a gene expression service using RNA in situ hybridisation or protein immunohistochemistry. Sectioned embryonic or fetal tissue is used to identify the temporal and spatial expression of specific genes or proteins. Gene expression data from this service, as well as data generated by external projects, are digitally imaged and made accessible via an open access database (www.hudsen.org). Some stained sections are also available at high resolution (http://nbb-slidepath.ncl.ac.uk/dih; username and password available on request).
Material can be provided to researchers in the UK without the need for project ethics review, as well as to international groups with relevant ethical review body approvals. Over 10,000 slides and tissue samples have been distributed to registered users in the last 5 years, and ∼100 projects are registered per year. The majority of projects are based in the UK, but a growing number of users are in the USA (17%), mainland Europe (11%) and other locations worldwide. Distribution of human embryonic and fetal tissue is prioritised to projects that investigate congenital disorders. Of particular interest are studies that aim to understand the function of genes important for early development, genes linked to human-specific functions (e.g. cognitive function and language) and genes associated with significant anatomical or functional differences between mice and humans.
A wide variety of studies have been carried out using HDBR materials (Fig. 2). These range from investigation of single genes underlying genetic disorders (Tischfield et al., 2010;Thomas et al., 2014) to high-throughput studies of transcriptomes (Kang et al., 2011) and regulatory sequences (Necsulea et al., 2014). Advances in high-throughput sequencing technologies have opened the door to large-scale cross-species comparisons that are highlighting differences in transcript sequence, alternative splicing, expression level and regulation between humans and model organisms (Cotney et al., 2013; Bae et al., 2014). Some studies, such as the investigation of tail bud development in chick and human (Olivera-Martinez et al., 2012), dissect key processes of early development. Organ-specific tissues have been used for the derivation of primary cell lines and stem cells (U et al., 2014).
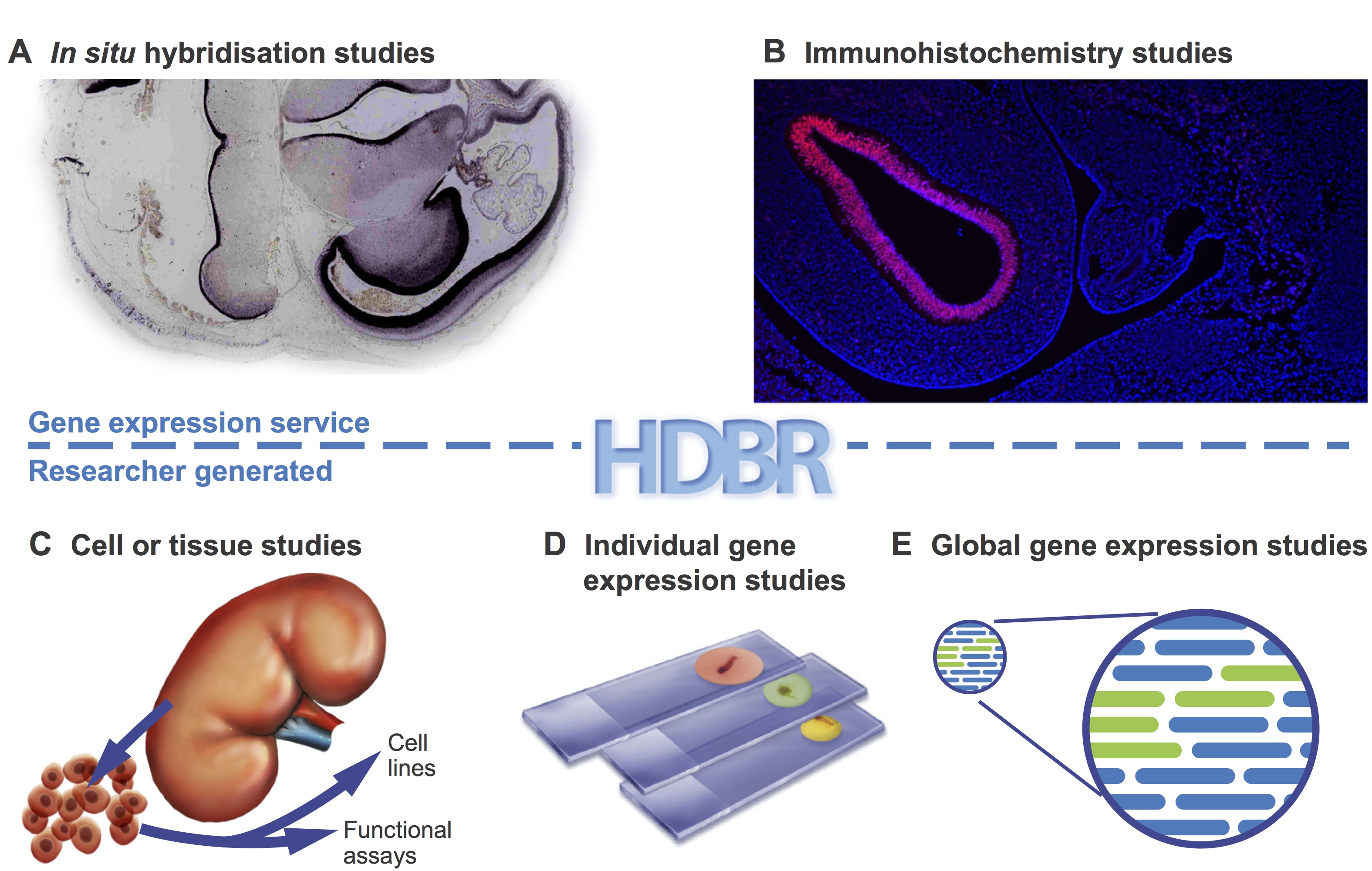
Fig 2. HDBR material has been used in many different types of studies.Examples are shown of the types of study carried out using HDBR material. (A,B) Examples from the gene expression service. HDBR staff perform gene expression analysis on sectioned tissue by in situ hybridisation (A) or immunohistochemistry (B) on behalf of researchers. (C-E) The types of studies that researchers carry out in their own laboratories with material provided by HDBR: to produce cell lines or perform functional analysis (C); gene expression studies in paraffin fixed sections (D); and transcriptomics studies in cells or tissues (E).
Other tissue banks
A number of other groups, in the UK and USA, have set up collections of human fetal material. These are normally organised around defined scientific projects and the samples are not usually available to the wider scientific community. However, there are exceptions where banks have been established to provide material for use in research. For example, the South Wales Initiative for Fetal Tissue (http://www.biobankswales.org.uk/swift-research-tissue-bank/) provides clinical grade fetal tissue (5 and 12 pcw) primarily for human therapeutics, and the University of Maryland Brain and Tissue Bank in the USA (http://medschool.umaryland.edu/btbank/catalog.asp) can provide access to both adult and fetal tissue.
Bioinformatics portals
A number of sites provide valuable information on human development for researchers. For example, the BrainSpan project (www.brainspan.org) has generated transcriptome data and gene expression data from multiple regions of the embryonic, fetal and adult brain. The UNSW Embryology portal is an education and research website that has links to many collections of human embryonic and fetal specimens (https://embryology.med.unsw.edu.au/embryology/index.php/).
In the UK a consortium of UKCRC funders has established The National Tissue Directory and Coordination Centre (www.biobankinguk.org). This directory will enable researchers to find human biobanks within the UK and gain access to their collections through one system. These and other bioinformatics databases are essential resources for researchers, particularly those whose access to primary tissue is limited.
Archive resources
The Carnegie collection (www.ehd.org/virtual-human-embryo/)
Seven thousand human embryos are stored in the Carnegie collection at the National Museum of Health and Medicine, Washington, D.C. This material was used to develop the comprehensive Carnegie staging system based on internal as well as external features. Carnegie staging (CS1-23) is now employed universally in human embryo research. Individual embryos throughout the embryonic period have been sectioned and, in an attempt to make the collection more accessible for research and teaching, digital images from a subset of these have been acquired. The images are labelled with standard anatomy terms to help with interpretation, or used to generate 3D models and 252 movies.
The Kyoto collection (http://bird.cac.med.kyoto-u.ac.jp/index_e.html)
This is the largest human embryo collection in the world, with over 44,000 specimens between CS7 and CS23. Maternal epidemiological data and detailed clinical information on the pregnancies were collected for each specimen. Five hundred normal and 500 abnormal embryos have been serially sectioned, and a further 1300 staged human embryos have been digitally imaged by magnetic resonance (MR) microscopy and 3D reconstructions produced.
Conclusions
The landscape for developmental biology and clinical research has changed within the last few years, with more emphasis being placed on large-scale sequencing projects such as the Genotype-Tissue Expression Project (GTEx Consortium, 2013), Geuvadis (Lappalainen et al., 2013) and the UK 100,000 Genomes Project (Siva, 2015). These projects and numerous others aim to map variations in gene expression in thousands of individual patients and correlate this with disease phenotypes and bioinformatics information on sites such as the Encyclopedia of DNA Elements (ENCODE; https://www.encodeproject.org/;Kellis et al., 2014). Other studies are using novel algorithms to interrogate the sequencing data to investigate gene networks (Liu et al., 2014). These projects are now identifying candidate genes that could play a role in a range of human diseases and syndromes.
The next step in this gene discovery pipeline will be to test candidate genes in model organisms and human tissue. This analysis needs to be performed at the cellular level to determine whether the genes are expressed in tissues and cell types relevant to the disease under study. The HDBR is ideally placed to assist researchers wishing to investigate genes thought to be responsible for congenital abnormalities. In addition, some groups are using material provided by the HDBR to study epigenetic regulation and in functional studies. Other projects include the derivation of stem cells and scaffolds for tissue engineering projects. It is likely in the coming years that the HDBR will not only support projects wishing to understand key developmental processes but will also increasingly provide resources to underpin translational research.
References
Bae, B.-I., Tietjen, I., Atabay, K. D., Evrony, G. D., Johnson, M. B., Asare, E., Wang, P. P., Murayama, A. Y., Im, K., Lisgo, S. N. et al. (2014). Evolutionarily dynamic alternative splicing of GPR56 regulates regional cerebral cortical patterning. Science 343, 764-768. doi:10.1126/science.1244392
Bae, B.-I., Jayaraman, D. and Walsh, C. A. (2015). Genetic changes shaping the human brain. Dev. Cell 32, 423-434. doi:10.1016/j.devcel.2015.01.035
Blaschke, R. J. and Rappold, G. (2006). The pseudoautosomal regions, SHOX and disease. Curr. Opin. Genet. Dev. 16, 233-239. doi:10.1016/j.gde.2006.04.004
Cadman, S. M., Kim, S.-H., Hu, Y., González-Martínez, D. and Bouloux, P.-M. (2007). Molecular pathogenesis of Kallmann’s syndrome. Horm. Res. 67, 231-242. doi:10.1159/000098156
Cotney, J., Leng, J., Yin, J., Reilly, S. K., DeMare, L. E., Emera, D., Ayoub, A. E., Rakic, P. and Noonan, J. P. (2013). The evolution of lineage-specific regulatory activities in the human embryonic limb. Cell 154, 185-196. doi:10.1016/j.cell.2013.05.056
Dolk, H., Loane, M. and Garne, E. (2010). The prevalence of congenital anomalies in Europe. Adv. Exp. Med. Biol. 686, 349-364. doi:10.1007/978-90-481-9485-8_20
Eichholzer, M., Tönz, O. and Zimmermann, R. (2006). Folic acid: a public-health challenge. Lancet 367, 1352-1361.doi:10.1016/S0140-6736(06)68582-6
Gautam, P., Anstey, K. J., Wen, W., Sachdev, P. S. and Cherbuin, N. (2015). Cortical gyrification and its relationships with cortical volume, cortical thickness, and cognitive performance in healthy mid-life adults. Behav. Brain Res. 287, 331-339. doi:10.1016/j.bbr.2015.03.018
GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580-585. doi:10.1038/ng.2653
Kang, H. J., Kawasawa, Y. I., Cheng, F., Zhu, Y., Xu, X., Li, M., Sousa, A. M. M., Pletikos, M., Meyer, K. A., Sedmak, G. et al. (2011). Spatio-temporal transcriptome of the human brain. Nature 478, 483-489. doi:10.1038/nature10523
Kellis, M., Wold, B., Snyder, M. P., Bernstein, B. E., Kundaje, A., Marinov, G. K., Ward, L. D., Birney, E., Crawford, G. E., Dekker, J. et al. (2014). Defining functional DNA elements in the human genome. Proc. Natl. Acad. Sci. USA 111, 6131-6138. doi:10.1073/pnas.1318948111
Lappalainen, T., Sammeth, M., Friedländer, M. R., t Hoen, P. A. C., Monlong, J., Rivas, M. A., Gonzàlez-Porta, M., Kurbatova, N., Griebel, T., Ferreira, P. G. et al. (2013). Transcriptome and genome sequencing uncovers functional variation in humans. Nature 501, 506-511. doi:10.1038/nature12531
Liu, L., Lei, J., Sanders, S. J., Willsey, A. J., Kou, Y., Cicek, A. E., Klei, L., Lu, C., He, X., Li, M. et al. (2014). DAWN: a framework to identify autism genes and subnetworks using gene expression and genetics. Mol. Autism 5, 22. doi:10.1186/2040-2392-5-22
Necsulea, A., Soumillon, M., Warnefors, M., Liechti, A., Daish, T., Zeller, U., Baker, J. C., Grützner, F. and Kaessmann, H. (2014). The evolution of lncRNA repertoires and expression patterns in tetrapods. Nature 505, 635-640. doi:10.1038/nature12943
Olivera-Martinez, I., Harada, H., Halley, P. A. and Storey, K. G. (2012). Loss of FGF-dependent mesoderm identity and rise of endogenous retinoid signalling determine cessation of body axis elongation. PLoS Biol. 10, e1001415.doi:10.1371/journal.pbio.1001415
Siva, N. (2015). UK gears up to decode 100,000 genomes from NHS patients. Lancet 385, 103-104. doi:10.1016/S0140-6736(14)62453-3
Thomas, M. G., Crosier, M., Lindsay, S., Kumar, A., Araki, M., Leroy, B. P., McLean, R. J., Sheth, V., Maconachie, G., Thomas, S. et al. (2014). Abnormal retinal development associated with FRMD7 mutations. Hum. Mol. Genet. 23, 4086-4093. doi:10.1093/hmg/ddu122
Tischfield, M. A., Baris, H. N., Wu, C., Rudolph, G., Van Maldergem, L., He, W., Chan, W.-M., Andrews, C., Demer, J. L., Robertson, R. L. et al. (2010). Human TUBB3 mutations perturb microtubule dynamics, kinesin interactions, and axon guidance. Cell 140, 74-87. doi:10.1016/j.cell.2009.12.011
Tookey, P. A. and Peckham, C. S. (1999). Surveillance of congenital rubella in Great Britain, 1971-96. BMJ 318, 769-770.doi:10.1136/bmj.318.7186.769
U, K. P., Subramanian, V., Nicholas, A. P., Thompson, P. R. and Ferretti, P. (2014). Modulation of calcium-induced cell death in human neural stem cells by the novel peptidylarginine deiminase–AIF pathway. Biochim. Biophys. Acta 1843, 1162-1171. doi:10.1016/j.bbamcr.2014.02.018
Yi, Y., Lindemann, M., Colligs, A. and Snowball, C. (2011). Economic burden of neural tube defects and impact of prevention with folic acid: a literature review. Eur. J. Pediatr. 170, 1391-1400.doi:10.1007/s00431-011-1492-8
Yue, F., Cheng, Y., Breschi, A., Vierstra, J., Wu, W., Ryba, T., Sandstrom, R., Ma, Z., Davis, C., Pope, B. D. et al. (2014). A comparative encyclopedia of DNA elements in the mouse genome. Nature 515, 355-364. doi:10.1038/nature13992
 (No Ratings Yet)
(No Ratings Yet) (6 votes)
(6 votes)
 (2 votes)
(2 votes) Here are the highlights from the
Here are the highlights from the  Gerrelli and colleagues summarise the remit and efforts of the Human Developmental Biology Resource – and other similar projects – that provides researchers with access to human tissue and related services. See the Spotlight article on p.
Gerrelli and colleagues summarise the remit and efforts of the Human Developmental Biology Resource – and other similar projects – that provides researchers with access to human tissue and related services. See the Spotlight article on p.  In their opinion piece, Thies and Murry consider progress in the use of human PSCs in regenerative medicine – fuelled by advances in developmental biology – in five areas that offer great promise for clinical applications. See the Spotlight article on p.
In their opinion piece, Thies and Murry consider progress in the use of human PSCs in regenerative medicine – fuelled by advances in developmental biology – in five areas that offer great promise for clinical applications. See the Spotlight article on p.  Vera and Studer discuss the difficulties involved in using stem cells to study diseases of the elderly and review how these might be overcome. See the Spotlight article on p.
Vera and Studer discuss the difficulties involved in using stem cells to study diseases of the elderly and review how these might be overcome. See the Spotlight article on p.  Pera and colleagues examine recent efforts in defining and capturing the human naïve pluripotent state in vivo and in vitro, in light of the different states of pluripotency found in the mouse. See the Review article on p.
Pera and colleagues examine recent efforts in defining and capturing the human naïve pluripotent state in vivo and in vitro, in light of the different states of pluripotency found in the mouse. See the Review article on p.  Franchini and Pollard discuss how genome sequencing and functional genomic approaches are enabling analyses of the evolutionary and developmental origin of traits unique to our species. See the Review article on p.
Franchini and Pollard discuss how genome sequencing and functional genomic approaches are enabling analyses of the evolutionary and developmental origin of traits unique to our species. See the Review article on p. 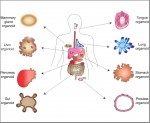 Huch and Koo review recent advances in the generation of ESC- and adult stem cell-derived organoids in order to understand the development of endoderm-derived organs in human and to develop therapeutic strategies for repair. See the Review article on p.
Huch and Koo review recent advances in the generation of ESC- and adult stem cell-derived organoids in order to understand the development of endoderm-derived organs in human and to develop therapeutic strategies for repair. See the Review article on p.  Jennings, Hanley and colleagues present a human-centric view of the latest advances in our understanding of pancreas development and the relevance of these insights from a clinical perspective. See the Review article on p.
Jennings, Hanley and colleagues present a human-centric view of the latest advances in our understanding of pancreas development and the relevance of these insights from a clinical perspective. See the Review article on p.  Suzuki and Vanderhaeghen examine stem cell-based approaches to analysing human brain development, from specification of particular cell types to building neuronal networks. See the Review article on p.
Suzuki and Vanderhaeghen examine stem cell-based approaches to analysing human brain development, from specification of particular cell types to building neuronal networks. See the Review article on p.