Looking back on the adventure: exploring cell fate conversion with single cell RNA-seq
Posted by Ziqing, on 21 November 2017
The story behind our recent paper: Liu Z*, Wang L*, Welch JD*, Ma H, Zhou Y, Vaseghi HR, Yu S, Wall JB, Alimohamadi S, Zheng M, Yin CY, Shen WN, Prins JF, Liu JD, Qian L (2017). Single cell transcriptomics reconstructs lineage conversion from fibroblast to cardiomyocyte. Nature, 551, 100-104 *: co-first author. https://www.nature.com/articles/nature24454
The Journey Started
In February 2014, I graduated from Indiana University with a PhD in Microbiology and Immunology. Being fascinated by regenerative medicine and how cell fate can be changed/ reversed during reprogramming, I joined Dr. Li Qian’s lab at University of North Carolina (UNC) in March 2014. Until today I still feel things just happened so fast and I am such a lucky person to be a part of this group. So I started my new journey at the beautiful small town of Chapel Hill to become a “reprogrammer.” Previously, Dr. Qian discovered that introduction of three cardiac transcription factors Mef2c, Gata4, and Tbx5 into mouse heart after myocardial infarction could significantly improve the heart’s function by converting the local fibroblasts in the heart to beating cardiomyocytes – which we termed as the process of “direct cardiac reprogramming” or “induced cardiomyocytes (iCM) reprogramming.” In addition to iCM, other researchers have reported the conversion of fibroblasts into induced neurons, hepatocytes, β cells, and so on. Direct lineage conversion not only offers a new strategy for tissue regeneration and disease modeling, but also provides a unique platform for understanding cell fate.
The Cell, the Single Cell
The cell is the most basic functional unit of an organism. The human body contains more than 40 trillion cells that are from different origins, form different organs, and carry out different functions. Each cell has its unique transcriptome, proteome, and functional role even though all cells originate from the same zygote and in theory share the same genome. Due to technical limitations, researchers were only able to check either the expression of limited number of genes at the single cell level (like immunofluorescence staining) or the transcriptome but at the price of averaging signals from a population of cells (like bulk RNA-sequencing). As the dawn of the era of single cell OMICs, we realized that we could do more now to understand the mechanisms of cell fate determination.
Direct Cardiac Reprogramming
Direct cardiac reprogramming shows promise as an approach to replenish lost cardiomyocytes in diseased hearts and its utilization of local scar-forming fibroblasts adds to its chance of potential clinical application. Considerable efforts have been made to improve the efficiency and unravel the underlying mechanism. However, it still remains unknown how the conversion of fibroblast into cardiomyocyte is achieved without following the conventional cardiomyocyte specification and differentiation. The reprogramming process is inherently heterogeneous in that the starting cells (primary fibroblasts) exhibit uncharacterized molecular heterogeneity and they don’t reprogram at the same pace, rendering it difficult to study using conventional bulk RNA-seq. Therefore, we decided that we were going to leverage the power of single-cell transcriptomics to really dissect the cellular and molecular mechanisms of iCM reprogramming.
Collaborator Hunting

Everything was perfect and exciting except that we had never done single cell RNA-seq at that time. Not many people around have either. So now, it’s time for the wonderful collaborators to come to the stage! First is the co-first author of the paper, my dear colleague and friend Dr. Li Wang, also from the Qian lab. This wonderful lady joined the lab earlier than me; she set up the experimental platform for everyone who joined after her and literally taught everyone hand-by-hand all the technical details to become a “reprogrammer”. This was our second project to co-first author. We designed and performed all the single cell experiments together. We discussed the results along the way and encouraged each other when things were not working. With the help of the UNC Advanced Analytics Core, we managed to get the experiments done with high quality. Nevertheless, throughout the time span of this project including the revision, we still went through difficulties such as changing the type and amount of control RNA spike-in due to evolvement of the technology and standard in the field, changing the version of the microfluidic chip because of a new release from Fluidigm, and revising our experimental design to take into account the difference in starting material (mRNA abundance) between different treatments.
Now we have the data! But how should we analyze it? In order to do data analysis, I took a great two-week workshop organized by UNC High Throughput Sequencing Facility that jumpstarted my bioinformatics and enabled some basic analyses by myself. But as we all understand, a two-week workshop and some self-learning are not sufficient for a high-quality paper and we needed a real expert at that time, desperately. So here is the magic how we found our wonderful collaborator, the computer modeling expert and the other co-first author of the paper, Dr. Josh D. Welch . To tell the story, I had to mention Haley Ruth Vaseghi first. Haley is a graduate student in the Qian lab. She knew that we were looking for help with single cell RNA-seq data analysis and her best girlfriend’s husband happened to be an computer modeling expert with a strong interest in analyzing single cell omics data. So she basically connected us by email and said:” Hey, maybe you will want to talk to each other~” Then we found our amazing collaborator Josh, who was still a graduate student in Dr. Jan Prins’s lab in the Computer Science Department at UNC at that time, now a postdoc at the Broad Institute and Assistant Professor-to be at University of Michigan. I knew immediately after our first meeting that Josh was not those graduate students that you say “em…” or ”fine.” He was one of those few graduate students that you say “wow!” His passion, expertise, and sparkling ideas turned out to tremendously help the project. From experimental design to data processing and normalization, then to data modeling and analysis and manuscript writing, his input to this project ensured the quality of data analysis in the paper.

Back to the Science
With everything ready, we explored the mechanisms of iCM reprogramming using single cell RNA-seq. We analyzed the transcriptome of 513 single mouse neonatal cardiac fibroblasts that undergo reprogramming for 3 days. Combined with computational modeling and experimental validation, here are what we found:
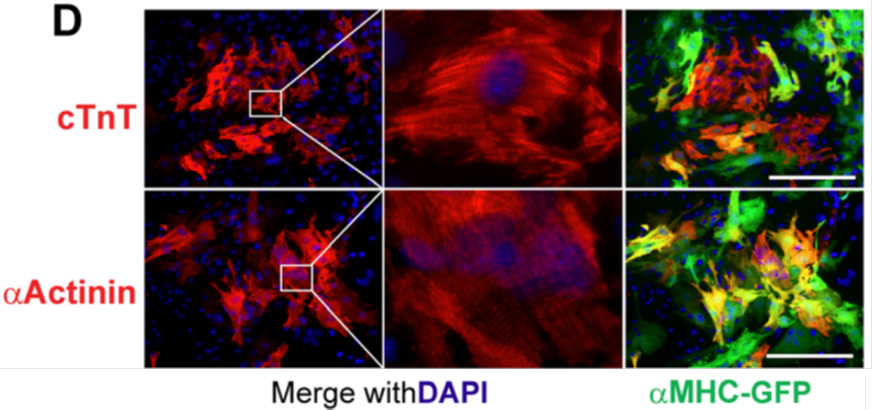
- Using unsupervised dimensionality reduction and clustering algorithms, we identified molecularly distinct subpopulations of cells during reprogramming, including an novel intermediate cell population pre-iCM that express both fibroblast (the start cell) and cardiomyocyte (the target cell) genes. Our findings here suggest that iCM reprogramming is different from the induced pluripotent stem cell reprogramming, which requires early down-regulation of fibroblast markers for reprogramming to proceed.
- We constructed the route of iCM formation using SLICER, an algorithm for inferring nonlinear cellular trajectories, and calculated pseudotime of reprogramming progress for each cell based on the trajectory. The trajectory revealed a bifurcation of reprogramming and proliferation states of cells.
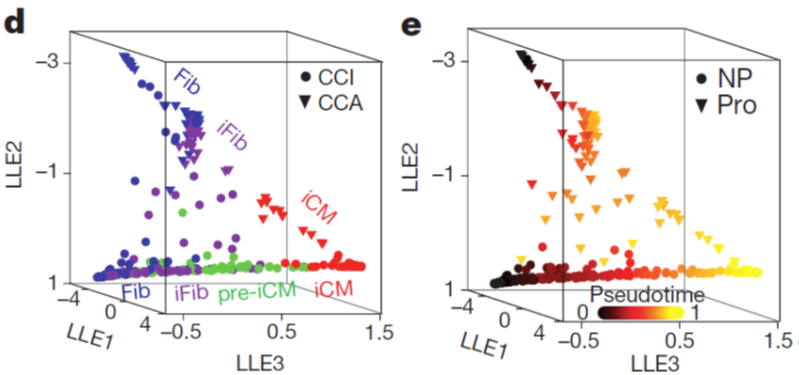
- We followed up on the trajectory and delineated the relationship between cell proliferation and iCM induction. Our data suggest that decreased proliferation or cell cycle synchronization promote iCM reprogramming and that increased proliferation inhibits reprogramming. This finding could be important for potential clinical application of reprogramming. Because after myocardial infarction, cardiac fibroblasts become activated and proliferative but their proliferation gradually decrease along with time post injury.
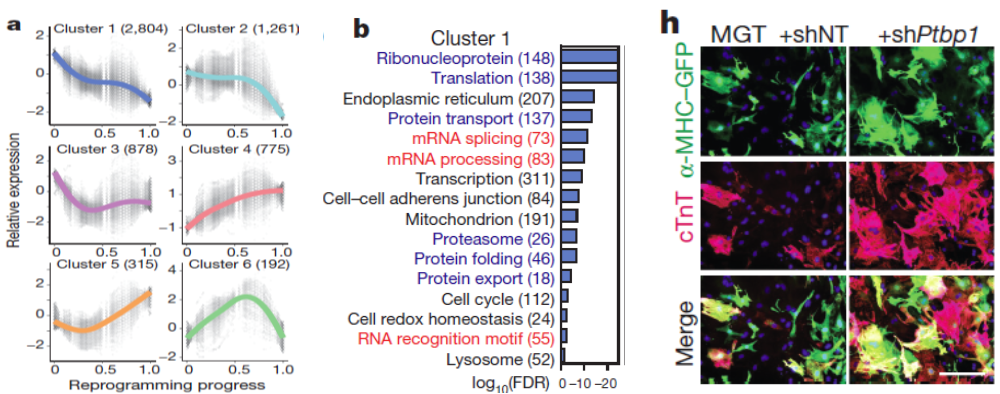
- We performed nonparametric regression and k-medoid clustering to identify clusters of genes that are significantly related to and show similar trends over the reprogramming process. We found immediate and continuous downregulation of genes involved in the basic cellular machineries of protein translation/ biosynthesis, modification and transportation. This is consistent with another observation during the data analysis that total mRNA abundance decreased by 40% upon reprogramming. These changes are probably to balance for increased energy requirements during the cell-fate switch and/or to transit from a protein production and ‘secretion factory’ (a fibroblast) to an energy-consuming ‘power station’ (a cardiomyocyte).
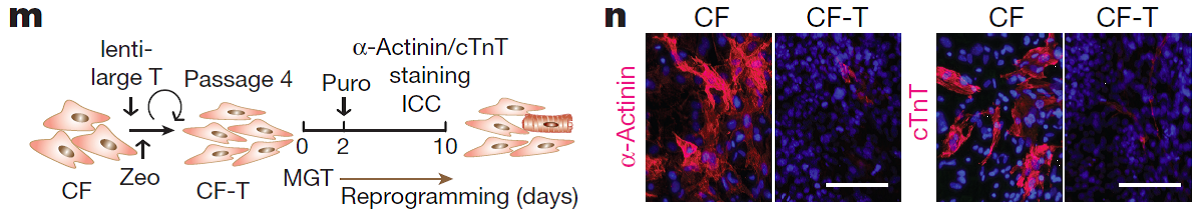
- Further analysis of global gene expression changes during reprogramming revealed unexpected downregulation of factors involved in mRNA processing and splicing. We therefore performed a loss-of-function screen against a library of major splicing factors and identified Ptbp1 as the top candidate. Detailed functional analysis revealed that Ptbp1 is a critical barrier for the acquisition of cardiomyocyte-specific splicing patterns in fibroblasts. Concomitantly, Ptbp1 depletion promoted cardiac transcriptome acquisition and increased iCM reprogramming efficiency.
- Additional quantitative analysis of our dataset revealed a strong correlation between the expression of each reprogramming factor and the progress of individual cells through the reprogramming process, suggesting that reprogramming is a process highly associated with/determined by the expression levels of reprogramming factors.
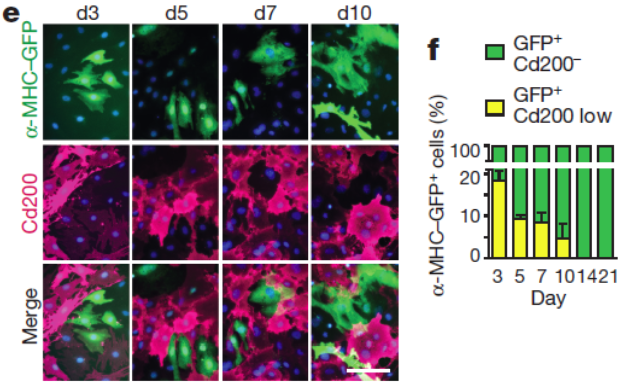
- Correlation analysis led to the discovery of new surface markers for the enrichment of iCMs such as the top negative selection marker candidate Cd200. Combinatorial use of these negative selection markers with positive selection cardiac reporters might enable convenient enrichment of iCMs using e.g., FACS sorting.
In summary, we used single-cell transcriptomics analysis to gain insights into the heterogeneity of cells within an unsynchronized cardiac reprogramming system. The findings show promise for improving the efficiency and detection of iCM formation. We also anticipate that the experimental and analytical methods presented here, when applied in additional cell programming or reprogramming contexts, will yield crucial insights into cell fate determination and the nature of cell type identity.
Everyone’s Efforts Make Things Happen— the Collaborative Qian Lab
Throughout this project especially during the revision process, everyone in the Qian lab and our collaborators all help a lot. In addition to the other two first authors mentioned above, Dr. Yang Zhou, another postdoc in the lab helped with a lot of the proliferation assays; Dr. Hong Ma helped with most of the Ptbp1-related experiments; Haley, as mentioned above, helped with the shRNA screen; our lab manager Dr. Chaoying Yin helped with cloning and a bunch of other things throughout the project; Dr. Weining Shen in University of California-Irvine, expert in stats and my college classmate helped with the statistical analysis; several other students and volunteers Blake, Sahar, Michael, and Shuo helped with different experiments. The two most important people behind this project are surely Dr. Jiandong Liu and Dr. Li Qian. They are not only a couple of passionate and talented scientists, but also caring mentors always supporting and guiding us on our way of scientific adventure. The most amazing part of my experience in the lab besides the quality and productivity of research is actually the unbelievable collaborative, encouraging, and happy environment Li and Jiandong have created. I think that is the exact reason why things can happen this way and everyone is enjoying both life and research in this lab. I feel blessed for being a part of the lab.


What Is Next?
Decades of accumulation of bioinformatics and computational modeling algorithms and improvement on sensitivity of each step in the sequencing pipeline finally led to the quantum leap that resulted in the advent of single cell OMICs. It is like the invention of smart phone – a completely new era of biological discovery has dawned. Single cell OMICs provide access to new biological questions or revisits of old biological questions but at an unprecedented resolution. Examples of questions to re-ask include “what defines a cell?”, “ultimately how is the fate of a cell determined?” and “what are molecular cascades to establish the cell fate once determined?”
In the world of big data, changes in technology and computer modeling algorithms are rapidly re-shaping the world around us and the way we do research. Interdisciplinary projects adopting combinatorial approaches of both biological experiments and computational analyses will open new windows of biological discoveries and inspire new ideas on both sides. In the future, we expect to continue this strategy of interdisciplinary collaboration and further explore cell fate control in development, regeneration, and disease.
 (5 votes)
(5 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.