Why more is better in comparative developmental biology…
Posted by Andreas Hejnol, on 26 January 2018
Our recent paper in “Nature” [1] deconstructs molecular arguments that have been used to homologize bilaterian nerve cords. Our work illustrates well the strength of the comparative approach and the broad sampling across the animal tree of life that we use in my research group at the Sars Centre for Marine Molecular Biology.
Evo-Devo branched off as a discipline from Developmental Biology. Therefore Evo-Devo inherited concepts and narratives from Developmental Biology that dominated over those of its cousin-discipline Evolutionary Biology. This was in part because Evo-Devo began with the comparison of data from a handful of well-studied genetic models, such as Drosophila, C. elegans, and vertebrates, which were used as a quite limited representation of the animal diversity. At this time, Evo-Devo was more “Devo” than “Evo”.
I entered Evo-Devo as a trained comparative zoologist who was –and still is– fascinated by animal diversity, evolution, and the many developmental pathways animals can display. I was attracted by the new discoveries of developmental biology, but being well familiar with the comparative method, I was also skeptical about their potential evolutionary implications, given the low taxon sampling (that is, the sparse number of species investigated across a tree).
At about the same time, molecular phylogenetic methods were shaking and uprooting the Animal Tree of Life. New molecular trees provided evidence for the monophyletic Ecdysozoa [2] (e.g. nematodes and arthropods), Lophotrochozoa [3] (e.g. annelids, molluscs) and Deuterostomia (e.g. urchins and frogs). Traditional relationships, such as the close relationship of annelids and arthropods, were questioned. However, the order of the internal branches remained unresolved. Such polytomies, together with the low taxon sampling by that time, obscured evolutionary conclusions about ancestral characters in these major groups, and thus hampered the detection of evolutionary change in the animal tree of life (Figure 1a). Instead, this situation sparked anthropogenic speculations.
Evo-Devo was thus influenced by two problems: the low number of studied species, and the lack of a phylogenetic framework that allows hypotheses testing.
This situation rapidly changed in areas of the Animal Tree of Life where taxonomical sampling was denser, and where the phylogenetic relationships were better resolved – such as in the vertebrates, and partly in the arthropods. This favored hypothesis testing, and improved our understanding of morphological evolution, as seen for instance with the evolution of limbs from fins [4]. But scientists interested in squishy invertebrates had much harder times.
Advances in sequencing technologies, imaging, and molecular methods have radically changed Evo-Devo, opening up new opportunities, and becoming a continuous source of innovation in evolutionary biology. Next generation sequencing has hugely impacted phylogenomics, allowing much better resolved animal relationships, thus providing the necessary phylogenetic frame to test evolutionary hypotheses [5]. These same sequencing technologies also make it now much easier to take animals from the field and implement molecular techniques faster for their study, which enables a much denser taxonomical sampling of animal diversity. And as sampling more data points provides a better measurement in physics, sampling more species provide a better understanding of evolution (Figure 1b). A denser sampling within a resolved phylogeny allows a more solid reconstruction of ancestral characters and offers a much better picture about character changes during evolution [6]. This approach is common practice in evolutionary biology [7] – but has not yet been extensively used in Evo-Devo.
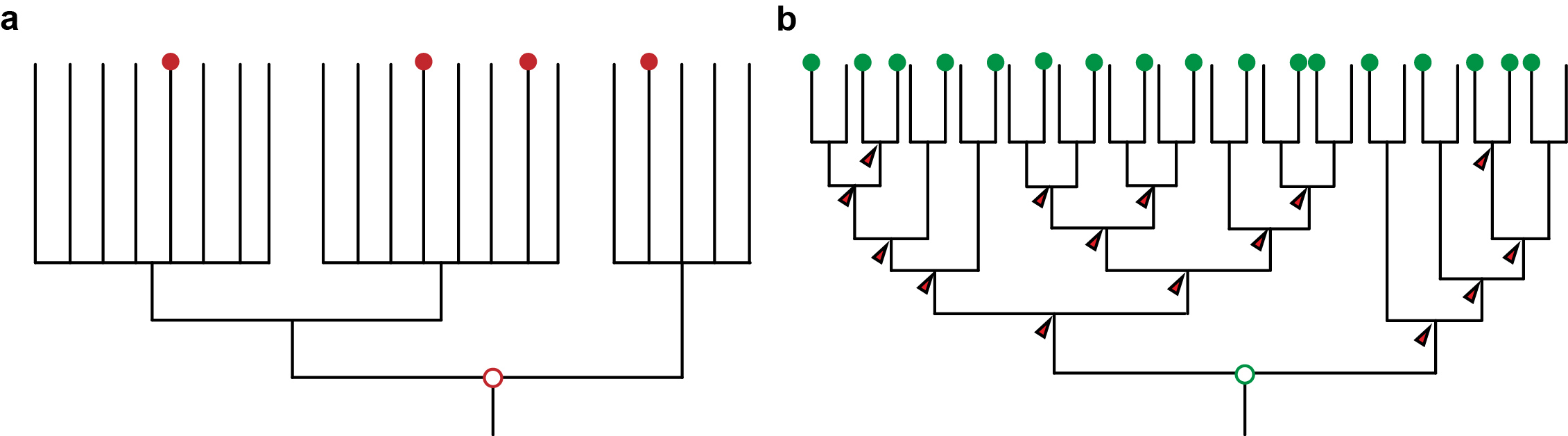
Figure 1: a) An unresolved phylogeny with only few taxa sampled (red dots) can only provide vague and untestable hypotheses. b) A well resolved phylogeny with large sampling allows the reconstruction at many more nodes and thus also insights about the evolution along the branches that connect the nodes. It also provides a framework for testing hypotheses.
The approach allows to test previous hypotheses that have been developed when low taxon sampling and/or unresolved phylogenies prevailed. For instance, our study of nine species published in “Nature” we show that the expression of the transcription factors traditionally used to call for a single condensed nerve cord in the last common ancestor of bilateral animals [8] evolved not only independently from each other, but also independently from the nervous system architecture and neuronal cell types. This implies that statements such as “this organ is so complex, it is unlikely to have evolved twice” are human presumptions, and that shared similarities might also be the result of independent adaptation processes (that is homoplasy). The comparative method strongly reduces human bias and also helps to avoid wrong conclusions [9].
The advantages of our approach not only extend to the study of organ system evolution, but simply affects all other characters that undergo evolutionary changes: tissues, cell types, developmental modes, genome architectures, gene expression [9]. Increasing the number of research organisms and the interpretation of the data on the basis of a resolved phylogeny provides a powerful toolkit for understanding evolution. It opens up the horizon for addressing new questions on when and how things changed in evolution, which is indeed where our curiosity lays.
References
- Martín-Durán JM, Pang K, Børve A, Semmler Lê H, Furu A, Cannon JT, et al. Convergent evolution of bilaterian nerve cords. Nature. 2018;553(7686):45-50.
- Aguinaldo AM, Turbeville JM, Linford LS, Rivera MC, Garey JR, Raff RA, et al. Evidence for a clade of nematodes, arthropods and other moulting animals. Nature. 1997;387(6632):489-93.
- Halanych KM, Bacheller JD, Aguinaldo AM, Liva SM, Hillis DM, Lake JA. Evidence from 18S ribosomal DNA that the lophophorates are protostome animals. Science. 1995;267(5204):1641-3.
- Shubin N, Tabin C, Carroll S. Fossils, genes and the evolution of animal limbs. Nature. 1997;388(6643):639-48.
- Dunn CW, Giribet G, Edgecombe GD, Hejnol A. Animal Phylogeny and Its Evolutionary Implications. Annu Rev Ecol Evol Syst. 2014;45:371-95.
- Hejnol A, Lowe CJ. Embracing the comparative approach: how robust phylogenies and broader developmental sampling impacts the understanding of nervous system evolution. Philos Trans R Soc Lond B Biol Sci. 2015;370(1684).
- Garamszegi L, editor. Modern Phylogenetic Comparative Methods and Their Application in Evolutionary Biology: Springer; 2014.
- Holland LZ, Carvalho JE, Escriva H, Laudet V, Schubert M, Shimeld SM, et al. Evolution of bilaterian central nervous systems: a single origin? Evodevo. 2013;4(1):27.
- Dunn CW, Zapata F, Munro C, Siebert S, Hejnol A. Pairwise comparisons across species are problematic when analyzing functional genomic data. Proceedings of the National Academy of Sciences of the United States of America. 2018;115(3):E409-E17.
Andreas Hejnol is Research Group Leader at the Sars International Centre for Marine Molecular Biology, University of Bergen, NORWAY. The group has currently vacant PhD and Postdoc positions.
 (8 votes)
(8 votes)Get involved
Create an account or log in to post your story on the Node.
Sign up for emails
Subscribe to our mailing lists.