C. elegans Development, Cell Biology and Gene Expression and 2018 European Worm Meeting report
Posted by Sophie PR Gilbert, on 9 July 2018
I recently attended the biennial Development, Cell Biology and Gene Expression C. elegans Meeting, this time in combination with the 2018 European Worm Meeting, in Barcelona. C. elegans meetings are always pretty special, defined more than anything else by the strong sense of community between researchers, or, as well like to call ourselves, ‘Worm People’. Worm culture has extended from a magazine (The Worm Breeder’s Gazette) to comedy shows (The Worm Show), and worm-inspired art collections (The Worm Art Show) curated by Ahna Skop, designer of this year’s meeting logo. This shared love of the worm translates into an atmosphere of supportiveness and openness within the community, making worm meetings ideal places to share new data and ideas – and at the same time being a huge amount of fun.
![]()
The meeting logo was designed by Ahna Skop to represent the extensive work that has been carried out investigating the neurobiology of the worm. C. elegans remains the only animal to have its entire connectome mapped.
A person who exemplified the ethos of the worm research community, and the memory of whom permeated the meeting, was John Sulston, who sadly passed away earlier this year. John Sulston was one of the first scientists working on C. elegans with Sydney Brenner at the LMB in Cambridge during the ’70s. He is known most famously within the worm field as the person who gave us the complete C. elegans cell lineage, a fundamental cornerstone of C. elegans research. This lineage represents years of work, as he monitored in real time every cell division of the larval – and following this, embryonic – worm, in order to build a remarkably accurate map of wild-type development from zygote to the adult. It is only very recently (40 years later) that any additional cell divisions have been identified. The concept of lineage is so fundamental to the ‘worm world’ that lab members also identify themselves according to their ‘lineage’ – tracing themselves back to that first ‘zygotic’ lab in Cambridge through a network of supervisors – in fact, many of the conference speakers introduced themselves in this way at the meeting.
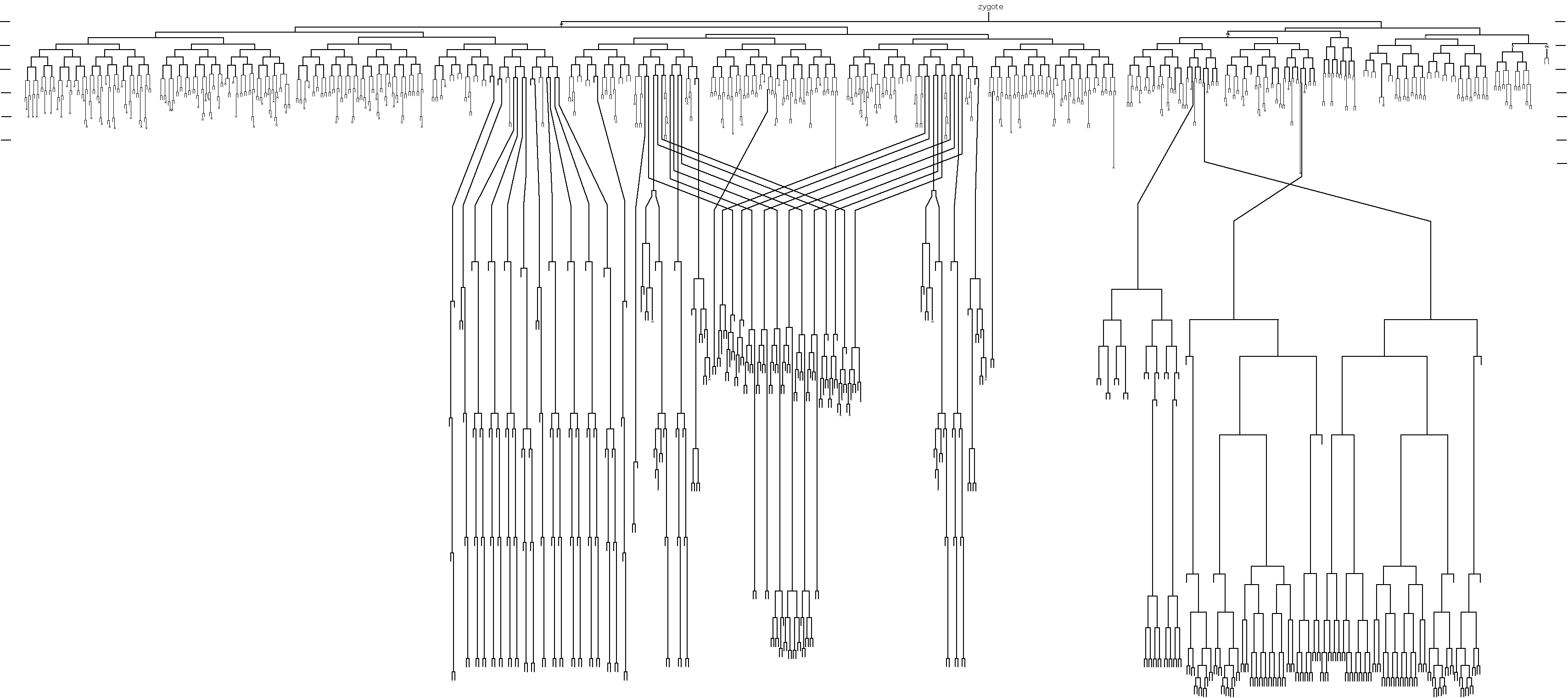
The C. elegans cell lineage, elucidated by John Sulston.
However, John Sulston’s scientific legacy extends beyond the relatively small sphere of C. elegans biologists, as he was also the scientist who lead the British side of the Human Genome Sequencing Project. A first consequence of this was that the C. elegans genome was actually completed first, cementing it’s place in history as the first sequenced genome of a multicellular organism (John was apparently just as excited – if not more so – to resolve the worm genome as to complete the human genome). But more importantly, John Sulston was also responsible for fighting to make the human genome sequence freely accessible to all, not patented and the property of a company. The contributions that John Sulston made to the scientific community during his time with the Human Genome Sequencing project, and how his work with C. elegans shaped his scientific ideology, can be read in a book called The Common Thread, which he wrote with the science writer Georgina Ferry. His attitude towards open-access science and the free sharing of data and resources remains a defining property of the C. elegans field.
The meeting: attendees and talks
The meeting, held at the World Trade Centre on the waterfront in Barcelona and organised by Sander van den Heuval, Sophie Jarriault and Alex Hajnal, opened to an audience of 465 people, representing research from 33 different countries. As well as the talks, the meeting also involved two lively poster sessions, and a fabulous evening of food, wine and dancing by the sea – and where better to party than in Spain? I certainly came away from the meeting with more worm-sympathetic friends than I’d started with.
As there were unfortunately too many presentations to talk about them all here, I have summarised a selection of my favourites. However, a complete list of the abstracts can be found here.

Meeting attendees, representing 33 countries.

The World Trade Centre (arrow) in Barcelona, where the meeting was held. Photo by the author.
Worm behaviour and neurogenesis
When Sydney Brenner first selected Caenorhabditis for research, his ultimate goal was to solve the genetics and cell biology of behaviour. To this end, the electron microscopist Nichol Thompson created serial sections (up to 5000, uninterrupted) of the worm in order to map each synapse, and subsequently a draft of ‘the Mind of a Worm’ was published in 1986. Given that the worm is the only organism to date to have it’s entire connectome mapped, the field of neurobiology – and more recently behaviour – is an intensely studied area of C. elegans research.
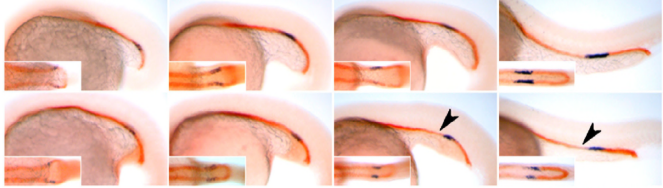
Upon completion of the C. elegans post-embryonic lineage, it was established that the adult hermaphrodite worm had 302 neurones, and the male 385. However, as I have already mentioned, the first post-Sulston cells to be identified added two neurones to the male count, discovered by Arantza Barrios of UCL, London, and published in 2015. At this meeting, Arantza Barrios revealed how her lab, together with the lab of Richard Poole, also at UCL, have since gone on to update the number of male neurones yet again (to 389) although this time the cells in question were always known to be there. However, they have now been identified as undergoing a fate and morphology change during larval development, producing a pair of putative proprioceptive neurones, the PHDs, from a pair of glial cells. The Barrios and Poole labs, with the help of Scott Emmons and Steven Cook at the Albert Einstein College of Medicine in New York, who through electron microscopy established the connectivity of the newly discovered neurones, have identified that the PHDs are essential for efficient male mating behaviour. As Arantza described, males are usually lead by the tail (in which their hermphrodite-sensing organ and spicule for sperm-delivery are located) during mating. In contrast to this known mating behaviour, a newly-described behaviour (termed the Molina Manoeuvre after the post-doc who discovered it) involves an unusual forward sweep along the hermaphrodite followed by a reversal in direction in order to relocate the vulva. This manoeuvre is only performed in the case of the male being unsuccessful in its first attempt to mate. Without the PHDs, the Molina Manoeuvre cannot take place successfully and the male is likely to lose its potential mate.
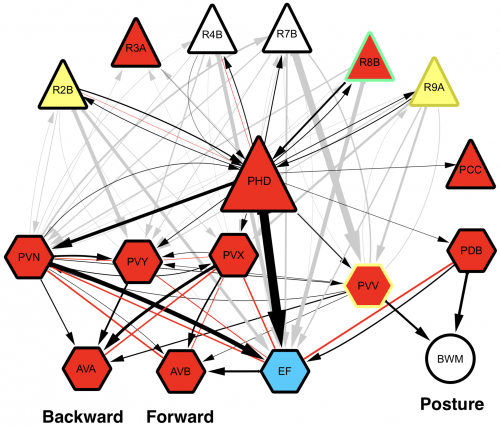
Wiring diagram of the newly-discovered PHD neurones, shown to be involved in male mating behaviour. L. Molina-Garcia et. al. “A direct glia-to-neuron natural transdifferentiation ensures nimble sensory-motor coordination of male mating behaviour” BioRxiv
Many mutant animals, or wild-type animals subjected to different environments, exhibit changed behavioural patterns. Systematic quantification of this behaviour can allow both detection of the behavioural differences and analysis of the specific change. Andre Brown of Imperial College London spoke about how his lab have broken down the 2-dimensional behaviour of C. elegans in a petri dish into a series of distinct positions, and uses the analogy of language to compare these positions to letters in an alphabet. He showed, using automated imaging of free-moving worm populations followed by extraction of relevant features, how wild-type worms are more likely to use a certain subset of ‘letters’ compared to mutant animals. His newly developed algorithms and imaging systems are likely to be useful for a wide range of applications, from behavioural genetics to agrochemical analysis.
Oliver Hobert from Columbia University in New York spoke about his lab’s recent feat of creating a comprehensive neurotransmitter map which overlays the anatomical map of the worm connectome, completed for both hermaphrodites and males. Most neurones in the worm fall into one of the main neurotransmitter classes: glutamatergic, GABAergic, or cholinergic. Upon completion of the neurotransmitter map, the Hobert lab has been able to identify 86 different classes of neurone within these classes, and through mining of the Wormbase database of gene expression, further show how a group of only 33 different transcription factors are able control these identities, acting as ‘terminal selectors’. (Remarkably, Oliver points out, the Wormbase collection of C. elegans gene expression patterns allows between 5 and 141 cell markers to be identified for each neurone – that’s an average of 32 markers per cell.) The Hobert lab deduced that there must, therefore, be a combinational code for neurone identity, and Oliver described experiments where the loss of one of a pair of transcription factors can have varying consequences on the identity of differing neurones. A second observation was that these terminal selector transcription factors are predominantly homeobox genes (c.f. only 10% of all transcription factors in the worm are homeobox genes). The lab went on to develop a worm strain that could be used to identify gene expression in any neurone based on co-expression (NeuroPal – Neuronal Polychromatic Atlas of Landmarks) and characterised gene expression of all homeobox genes in all neurones. They found that every neurone expresses at least one homeobox gene, with 113 out of 118 neurone classes expressing a unique homeobox gene code. This means the same homeobox gene gets used again and again to specify different neurones. A striking observation was made when looking for commonalities between the neurones that express the same transcription factor: they are all synaptically connected. This has lead to speculation in the lab of how evolution may first have resulted in individual networks of neurones, each based on the presence of a founding homeobox gene specifying a single neurone that subsequently duplicated, before acquiring additional specialisation across the network to encode multiple neuronal types. These networks, Oliver proposes, may have later linked up to form the connectome as we know it.
Tissue-specific expression analyses
Tissue-specific RNA-seq in the worm has provided a challenge for the field, especially for those interested in understanding the factors that drive differentiation. Current analyses include using known gene expression profiles coupled with machine learning algorithms to sort sequencing profiles obtained from FACS-sorted single cells back to their tissue of origin, such as the efforts of John Murray’s laboratory at the University of Pennsylvania. John presented his lab’s work on dissecting the cell-specific embryonic expression profile over time, using a ‘whole-organism shotgun’ approach. This approach involves using the RNA-seq data obtained from individual, separated cells from mixed stage embryos. He then profiles each cell in order to map it back to the cell lineage, using known gene expression ‘hallmarks’ obtained from expression patterns of individual genes. Using this data, it is possible to start to answer questions such as which genes are required for acquiring cell identity versus those that are required for maintaining cell identity. An interactive database of this data is under development and should be available soon (keep an eye on BioRxiv). In contrast, Annabel Ebbing, from Hendrik Korswagen’s lab at the Hubrecht Institute in the Netherlands, described how RNA tomography can contribute to our understanding of spatially-resolved gene expression patterns without any prior knowledge of gene expression – rather, this approach relies on the highly invariable anatomy of the worm. RNA tomography involves cryo-sectioning young adult animals at 20µm intervals along the anteroposterior axis, before using CEL-seq to sequence each section. The data were aligned to provide a comprehensive map of gene expression along the anteroposterior axis of the worm, at single cell resolution. This map has already revealed sex-specific differences in the expression of previously-unidentified genes required for male fertility in the male reproductive tract, and has been made available here.
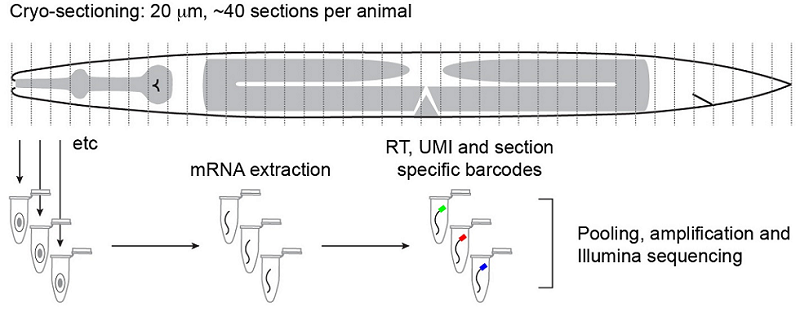
Outline of the RNA tomography method. Worms are sectioned at 20µm intervals before RNA from individual sections is sequenced. The relatively simple anatomy of the worm allows for single-cell and tissue resolution of sequencing data using this section width. Ebbing, A.*, Vertesy, A.*, Betist, M., Spanjaard, B., Junker, J.P., Berezikov, E., van Oudenaarden, A., and Korswagen, H.C. Spatially-resolved transcriptomics of C. elegans males and hermaphrodites identifies novel fertility genes. Submitted.
Cell fate and plasticity
The understanding of cell fate determination, and its opposite, fate plasticity, is important due to the potential implications of reprogramming in cancers and regenerative medicine. C. elegans provides two models with which to study in vivo reprogramming: cells which undergo natural transdifferentiation, and induced conversion of cells by ectopically expressed transcription factors.
The most famous natural transdifferentiation event in the worm is the Y to PDA transition in the tail. Y is a fully differentiated rectal epithelial cell that, partway through larval development, detaches from the rectum, migrates and converts into the PDA neurone. This process was first detected by John White and noted in his lineage, and later confirmed to be a bona fide transdifferentiation event by Sophie Jarriault. Sophie’s lab have continued to contribute to the field of reprogramming, and more recently her work has expanded to encompass other natural fate changes in C. elegans. Claudia Riva from the Jarriault lab talked about her work on these other cells. Although direct conversion of a cell from one fate to another is rare (the two known examples are the changing fate of Y, and also the phasmid socket PHso1, which produces the PHD neurone required for the Molina Manoeuvre), there are also cases of seemingly differentiated cells dividing to produce a cell of a different fate. These include the K cell in the rectum, which divides to produce the DVB neurone, and the G1 excretory pore cell, which gives rise to two RMH neurones. Claudia showed that even though these cells undergo a cell division, the ‘plasticity cassette’ comprising SEM-4, EGL-27, SOX-2 and CEH-6, identified to be required for the Y to PDA transition, is also required for the production of DVB from K. For example, in sem-4 mutant animals K goes on to divide but K.p remains epithelial. However, upon halting the cell cycle using a temperature sensitive allele of lin-5, she demonstrated that cell division is also essential for the production of DVB. The asymmetric division of K requires Wnt signalling, like many other asymmetric pathways in C. elegans, leading the lab to investigate the interplay of Wnt (during asymmetric cell division) and plasticity reprogramming factors (for the acquisition of neuronal cell fate) in the process of K dividing to produce the DVB.
Anna Reid, who works in the Tursun lab at the Berlin Institute for Medical Systems Biology in Germany, talked about her work on the forced reprogramming of mesodermal coelomocytes. There are three pairs of coelomocytes located in the pseudocoelom of the worm. The lab has found that the ectopic expression of the GATA factor ELT-2 is sufficient to convert these cells to the intestinal fate, and upon closer inspection found that they even produced an intracellular lumen lined with microvilli. Testing the limits of forced reprogramming, she found that the zinc transcription factor CHE-1, which specifies the fate of the ASE neurones, was also found to be able to reprogram the coelomocytes, with a 70% success rate for one of the three pairs of cells. Intriguingly, since the coelomocyte fate is lost at the time of conversion, so too is the expression of the coelomocyte-specific transgene inducing the forced transition event (in this case, the unc-22 promoter is used to drive ectopic che-1 expression in the coelomocytes). Thus only a pulse of expression must be sufficient to convert fate. Anna finds that this pulse of ectopic CHE-1 expression leads to the endogenous che-1 locus to be switched on in these cells; the fate-converting experiment in a che-1 mutant background is much less efficient. Lastly, Anna explained how the coelomocytes, now shown to be amenable to either intestinal or neuronal reprogramming, are much more resistant to a muscle fate change. Anna suggests that this is surprising, since the muscle lineage is much closer to that of the coelomocytes than the neuronal lineages. This raises the question of how lineage proximity can affect fate plasticity: if two lineages are closer, does this make it easier or harder to reprogram between them?
Transcriptional metabolic network rewiring
Marian Walhout, from the University of Massachusetts Medical School, USA, delivered a very elegant talk on the work her lab are carrying out in the field of systems biology. She explained how the worm has two metabolic pathways for breaking down proprionate, one of which is dependent on vitamin B12, and the other not. The B12-dependent pathway is preferable to the worm as it does not risk the build up of toxic intermediates, but the pathways are interchangeable in the event of no B12 being available and subsequent build up of proprionate. (Marian also points out that the standard laboratory culture conditions of growing C. elegans on the OP50 strain of E. coli means that the worms are always B12-deficient in the lab.) To convert from a B12-dependent pathway to a B12-independent pathway requires the transcriptional machinery to switch to expressing the enzymes required for the new pathway. However, in order to achieve this only when there is a sustained deficiency of B12, and not just in the case of a temporary peak in proprionate, requires a delay to be built into the switch. Marian explains how this delay has been modelled in bacterial systems by a coherent type 1 feed-forward loop with an AND-logic gate, providing persistence detection in response to a stimulus. However, this type of transcriptional circuitry has never been observed in an animal system, until now. To turn on the B12-independent pathway, Marian’s lab have found that two genes, nhr-10 and nhr-68, together act in such a persistence switch to turn on five genes of the B12-independent metabolic pathway, and in this way prevent this potentially hazardous pathway being turned on during a transient peak in proprionate.
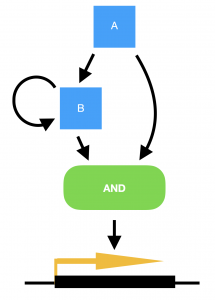
Model for a persistence detector in the form of a coherent type 1 feed-forward loop with an AND-logic gate. Transcription factors A and B are both required to turn on their targets. In the case of a transient stimulus, however, gene A will not be expressed long enough to make sufficient amounts of gene B, in order to turn on the switch. Yet if the stimulus is sustained, transcription factor A will activate the expression of gene B, and the target genes will be transcribed.
Meiosis
Abby Dernburg from the University of California, Berkeley shared her work on how crossovers are regulated during meiosis. In C. elegans, each pair of homologous chromosomes undergoes just one crossover event, and all other double-stranded breaks are repaired without the transfer of genetic information. Little is known about how chromosomes convey this crossover information along their lengths. Through fluorescent analysis, allowing direct visualisation of the synaptonemal complex assembly which forms between homologous chromosomes, the Dernburg lab have been able to quantify the kinetics of synapsis. This has lead to the conclusion that the synaptonemal complex in fact acts as a liquid crystal – an ordered assembly of molecules that show liquid-like mobility – rather than a static complex. This rapid diffusion within the complex could be the mechanism by which information is rapidly transported down the length of the homologous chromosomes. The lab is now testing how each of the previously-identified factors for regulating crossover events, including the RING finger proteins ZHP-1-4, CDK-2 and COSA-1, can act with the synaptonemal complex to regulate and limit the number of crossovers to one per chromosome pair.
Membranes in biology
David Sherwood of Duke University, USA, delivered a wonderful talk on his work studying basement membranes in C. elegans. I had first come across David’s work as a Masters student, reading about how the anchor cell invades through the basement membrane that separates the gonad from the uterine seam during vulval formation. During his talk, David described how his lab have created a toolkit of fluorescently labelled endogenous basement membrane components (17 components and 13 receptors) in order to study the composition and dynamics of this ancient extracellular matrix. One of the striking results from this work has been the discovery of the huge range in the rates of turnover and mobility between these tagged components: FRAP and FLIP experiments show that whereas some components take hours to recover, others recover in a matter of milliseconds.
Meera Sundaram from the University of Pennsylvania, USA, is more interested in the apical membranes of cells – during this meeting she shared some of the work from her lab on how the worm builds tiny, seamless tubes from a single cell. There are three possible ways for a single cell to form a tube: through endocytosis and internal vesicle fusion, through membrane invagination followed by exocytosis, or through auto-fusion after the cell has rolled around the lumen. In the excretory system of C. elegans, there are two cells which each form a tube. These are the duct cell, which forms a seamless tube, and the pore cell, which remains seamed. Both tubes are formed by an initial rolling of the cell, but the duct cell then auto-fuses in an EGF-Ras-ERK dependent manner. When looking for targets of this pathway, Meera’s lab discovered that this fusion is dependent on the activation of the fusogen aff-1, which has already been shown to drive fusion in the uterine seam and between seam cells. Auto-fusion of the duct cell occurs early in development, and is followed by morphogenesis and elongation of the tubular cell into its final shape. Using the degron system to deplete aff-1 after the fusion event but before remodelling, Meera showed that surprisingly, aff-1 was also required for the elongation of the duct cell. In the absence of aff-1, she showed using STED microscopy that the cell accumulates vesicles containing apical cargoes, and also had nucleus-sized ‘hair-balls’ of membrane attached to the basement membrane, suggesting that AFF-1 is required for apically-directed trafficking. AFF-1 is present at endocytosing membranes, so may be required for scission. Vesicle trafficking is known to be required for remodelling of the duct cell, as Rab11, required for the recycling of endocytic vesicles, is essential for duct cell elongation. Thus Meera’s lab has discovered a novel role for fusogens during vesicular trafficking.
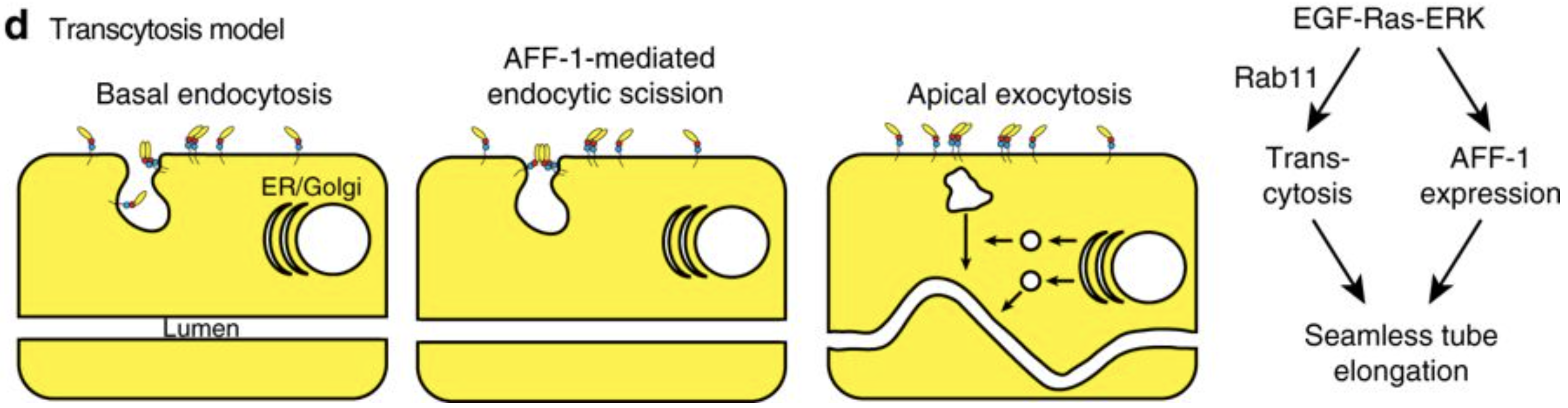
Model of the role of AFF-1 during endocytic scission in the duct cell of the excretory system. Soulavie F, Hall DH, Sundaram MV. The AFF-1 exoplasmic fusogen is required for endocytic scission and seamless tube elongation. Nature Communications. 2018;9:1741. doi:10.1038/s41467-018-04091-1
This was not the only talk at the meeting that presented a novel role for fusogens, however. Piya Ghose from the Shaham laboratory in the Rockefeller University, New York presented her work on how the fusogen EFF-1 was required for phagosome sealing during the programmed cell death of the tail spike cell. The role of fusogens in cell biology appears to be broader than was previously thought.
The ageing worm
The striking genetic control of worm lifespan has been demonstrated many times over the last few decades, and this together with the worm’s relatively short natural lifespan makes it an attractive model for ageing research. Adam Antebi of the Max Plank Institute for Biology of Ageing in Cologne, Germany, presented his lab’s work on how nucleolar size in the young worm can be used as a predictor of the lifespan of that worm – with worms with smaller nucleoli living the longest. Not only is this trend true for worms, but he showed how it also holds true for fruit flies, mice and humans, suggesting that the pathways of ageing that converge on nucleolar size are conserved. Adam’s lab found that nucleolar size is controlled by the NCL-1 tumour suppressor, with long-lived animals representing different longevity pathways exhibiting small nucleoli, decreased rRNA and ribosomal protein expression, and decreased fibrillarin, a nucleolar protein. Indeed, knockdown of fibrillarin is sufficient to reduce nucleoli size and increase lifespan in worms.
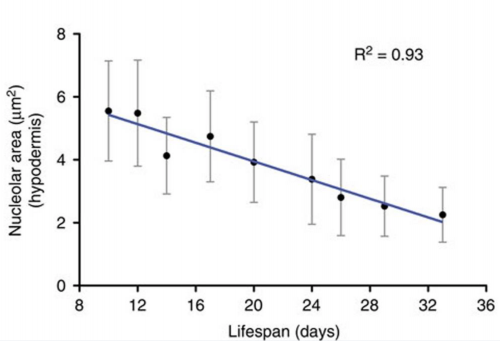
Nucleolar size inversely correlates with lifespan, and as such is a cellular hallmark of longevity. Tiku V, Jain C, Raz Y, et al. Small nucleoli are a cellular hallmark of longevity. Nature Communications. 2017;8:16083. doi:10.1038/ncomms16083.
Wild C. elegans
C. elegans research has primarily been performed on a single, Bristol isolate of C. elegans, called ‘N2’. However, there are myriad natural isolates of C. elegans around the globe. Resources for wild isolate research include those of Mark Blaxters lab in Edinburgh, and Erik Anderson, who manages the association mapping resource CeNDR. Jan Kemmenga has also produced a library of sequenced RILs between two highly polymorphic strains, the Bristol and Hawaiian isolates, that can be used to map differences in any phenotypic differences between these strains.
Marianne Félix, of the Marie Curie Institute in Paris, has been sampling wild C. elegans for many years, studying natural variation and using polymorphic genomes for quantitive genetic approaches to C. elegans research. Marianne described at the meeting a recently accepted (Lise Frézal et. al., Current Biology, in press) story investigating germline mortality in a C. elegans wild isolate. She described how the project started by leaving wild worms in a petri dish on her bench – resulting after a few generations in a sterile population. Luckily the lab was able to recover the population before it was too late, and realised they were observing a temperature-sensitive Mrt phenotype (mortal germline). Mrt phenotypes had already been observed in screens by Jonathan Hodgkin, which isolated mutants in DNA repair. The lab found that many of the wild isolates exhibited Mrt phenotypes to differing degrees of severity – the strain QX1211, for example, exhibits sterility in one generation. Many of these strains were isolated from hot countries, raising the question of how these strain survive in their natural habitats. Marianne observed that infection of the worms by natural pathogens was able to suppress the Mrt phenotype, indicating that the phenotype was in fact an artefact of the worms’ sterile environment in the lab petri dish. In order to understand the genetics behind why some strains exhibit the Mrt phenotype and not others, RILs were created between two strains, one Mrt and one not, JU1395 and MY10. Through phenotyping followed by whole genome sequencing, the Mrt phenotype was mapped to the set-24 locus on chromosome II. However, upon inspection of set-24 alleles across various wild isolates, Marianne’s lab found that the set-24 Mrt allele was, in fact, a rare allele in the wild. Thus to understand the common factor shared between the Mrt wild isolates, GWAS (genome-wide association mapping) was performed across 95 wild isolate genomes. This approach identified the more commonly present Mrt factor as morc-1, on chromosome III, a gene which helps to silence repetitive elements. Thus two different quantitive genetic approaches to find the cause of the same phenotype resulted in the identification of two different Mrt alleles, one of which was relevant for a particular strain (set-24), and one of which helped describe the differences between many wild isolates (morc-1).
Family, friends and enemies of Caenorhabditis
Although Caenorhabditis elegans took centre stage at the meeting, there were a few friends and even enemies of our favourite worm present. Marie Delattre from the Université de Lyon in France told us about one such worm, Mesorhabditis belari, which has a curious mechanism of genetic reproduction. For comparison, C. elegans has two sexes: hermaphrodite (of genotype XX) and male (of genotype X0). Populations are predominantly hermaphrodite, although the loss of one X chromosome at a rate of 1:1000 means that males do occur at a very low incidence. If a male mates with a hermaphrodite, mendelian genetics dictates that 50% of the resulting cross progeny, fertilised by sperm not carrying an X chromosome, are male. By contrast, Mesorhabditis belari is present in populations comprised of female and male individuals (so far so good). However, although the male is required by the female to fertilise her oocytes and to produce viable offspring, the male animal only rarely passes any of its DNA to the next generation, and progeny are predominantly clones of the female parent. So what is the point of producing males at all? Marie discovered that females produce two different kinds of embryos upon oocyte fertilisation. To understand the difference between them, it should first be mentioned that C. elegans oocytes, as well as those of Mesorhabditis belari, undergo only meiosis I before fertilisation by the sperm; meiosis II occurs shortly after fertilisation and it is only after expulsion of the sister gamete that the male pronucleus joins the female pronucleus to form the zygote. So, to go back to the oocytes of Mesorhabditis belari, it was found that 10% of oocytes carry out meiosis II after fertilisation, as expected, followed by fusion of the male and female pronuclei and subsequent development of the embryo – into male worms only. The remaining 90% of fertilised oocytes of Mesorhabditis belari do not complete meiosis II at all. Instead, the male pronucleus is prevented from expanding and fusing with the female nucleus, which enters mitosis to produce a female embryo – containing only the DNA of the female parent. In this scenario, it would seem that there is no evolutionary advantage to producing males. Marie turned to evolutionary game theory to provide some answers. She modelled two genetically distinct populations of Mesorhabditis belari, using factors to describe population size (i.e. how many progeny are produced) and how many worms will migrate away and towards each population. Using this model, she was able to identify a scenario in which males of Mesorhabditis belari presented an advantage to the population: if the males were to preferentially mate with their sisters. Indeed, Marie realised that the very low mating efficiency of these worms in the lab may also be explained by this preference, and upon performing the experiment, she found this to be the case. Thus, males provide their genetically similar sisters with sperm for the activation of oocytes, initiation of polarisation and for providing centrosomes – just not for their DNA.
A more nefarious creature (at least from the point of view of C. elegans researchers) is Pristionchus Pacificus. This is a dimorphous worm that in one of its forms is carnivorous – enjoying munching on C. elegans, amongst its other prey.
If one can overcome the macabre, however, an interesting question presents itself: how does Pristionchus Pacificus avoid becoming cannibalistic? James Lightfoot, from the lab of Ralf Sommer at the Max Planck Institute for Developmental Biology in Germany, explained how self-recognition (and thus avoidance of eating its own species) was achieved. In order to test the limits of self-recognition, James mixed populations of four different carnivorous species (including the sister species of Pristionchus pacificus) together on a plate, and observed that all four species recognised and avoided eating only themselves. If larvae from their own species were allowed to populate a plate and were later removed and replaced by the larvae of an alternative species, the new larvae were not protected from the predatory worms. Together these experiments suggested that self-recognition involved a surface-bound molecule. During his investigation, James found that individual isolates of Pristionchus pacificus, such as those isolated from Washington and those from California, will happily eat each other (even though they are members of the same species) but avoid worms from their own genetic pool. This observation allowed him to cross together the two strains to produce recombinant inbred lines (RILs), followed by phenotyping and sequencing the resulting strains. Impressively, the lab had to overcome a significant incompatibility region that obscured the data, and eventually managed this by inducing double stranded breaks along the chromosomes using CRISPR. This resulted in the identification of the locus ‘self-1’. Deletion of this locus resulted in a loss of self-recognition, and it was found to encode a 63 amino acid short peptide. An alignment of 38 strains revealed that although most of the self-1 sequence was highly conserved, it contained a highly variable terminal sequence. In addition, the copy number of this gene varied extensively between strains. James and his colleagues found that changing this variable region between strains was enough to protect the opposite strain, and that even a single amino acid change in this region was enough to affect self-recognition. However, he has identified some strains that carry identical self-1 sequences, but do not eat each other, suggesting that there are as yet still undiscovered mechanisms that regulate self-recognition in Pristionchus pacificus.
EMBO Young Investigator lecture
Luisa Cochella of the Research Institute of Molecular Pathology (IMP) in Vienna, delivered the EMBO Young Investigator lecture. Luisa studies the role of miRNAs (short, approximately 22nt long non-coding RNAs that repress gene expression) during development, and started her own lab after working as a postdoc in Oliver Hobert’s lab at Columbia University.

Luisa Cochella presents the EMBO Young Investigator lecture at the European Worm Meeting. Her slide is showing John Sulston’s lineage of the worm.
Luisa described how it has been previously proposed that miRNAs can act as ‘de-noisers’ of transcription, expressed in domains where their target genes are not normally transcribed, to reinforce the transcriptional control of the gene. Alternatively (and she suggests, perhaps more interestingly), miRNAs can act on their own to change cell identity. miRNAs begin their existence as miRNA precursor molecules (a self-annealing loop of RNA) which must be processed by first the RNase III Drosha, together with Pasha (partner of Drosha), and then again by Dicer to produce the mature miRNA which can be recognised by Argonaute. Embryos of a strain containing a temperature sensitive allele of Pasha, pash-1(mj100), fail to elongate during embryogenesis at the restrictive temperature, showing that miRNAs are essential for embryogenesis in the worm. Individual knockouts of 90 of a total 150 miRNAs, performed by Eric Miska and Bob Horwitz, showed no developmental arrest. However, upon deletion of entire miRNA families, they found that two families in particular were important for development. These were the mir-35 family (comprising 8 members) and the mir-51 family (6 members). Deletion of the mir-35 family phenocopies the pash-1 allele, whereas deletion of the mir-51 family resulted in animals that arrested slightly later during embryogenesis. Rescue of a miRNA gene family knockout strain can be achieved by any one of the family members.
By sequencing miRNA from embryos, Luisa revealed that 75% of all miRNA molecules belonged to one of the mir-35 or mir-51 families. Using a micro-processor by-pass strategy, which uses splicing machinery rather than Dicer to process synthetic ‘mirtrons’, the lab was able to rescue pash-1 mutant embryos with only two mirtrons, representing each of the mir-35 or mir-51 families. However, these animals still arrested during larval development, suggesting a role for the remaining 25% (by expression) of miRNAs. Despite some of these 25% of miRNAs being expressed at very low levels, Luisa hypothesised that they may be essential for very specific developmental events, and could be expressed in as little as one cell during development. She tested the expression patterns of these lowly-expressed miRNAs and found that they were, indeed, expressed in very restricted domains. One such miRNA, mir-791, was found to be expressed in a subset of neurones: BAG, AFD and ASE. These are all CO2-sensing neurones. In behavioural assays, loss of mir-791 resulted in a reduced avoidance response, and could be rescued by reintroduction of mir-791. This indicated that mir-791 has a specific role in conferring fate of these neurones, and is not merely involved in the robustness of the expression of target genes. Luisa found that the target genes for mir-791 were two ubiquitously expressed genes, akap-1 and cah-3. The only cells where the expression of these genes is repressed is in the CO2-sensing neurones. In this way, she has found that miRNAs may act in parallel to transcription factor control of gene expression to confer specific cell fates.
Keynote talk
Paul Sternberg from CalTech in California delivered the keynote talk at the meeting. The worm community owes Paul a huge debt for the many research resources that he has cofounded in his time working on C. elegans – including Wormbook, Textpresso and Wormbase. He began his career with C. elegans with the Horwitz lab at MIT at about the same time as John Suston’s lineage was published, at the start of what would become a hat tick for C. elegans research: the lineage, the connectome and the genome. Using this information, he set out to ‘solve the worm’. His lab maintain a very broad spectrum of research interests, and here he highlighted their recent work on the dauer life-form of the worm. C. elegans larvae enter an alternative lifecycle during conditions of starvation or overcrowding, in which they may remain as a dauer animal for up to three months. Indeed, in the wild this is the most commonly-found life stage. Upon finding food, the dauer animals re-enter the life cycle as L4 larvae, and progress to adulthood. Paul suggests that the dauer stage might also serve other functions in the worm, however, than just a means to survive starvation. He notes that animals of the genus Steinernema are able to jump significant distances as dauers, due to the energy stored in their cuticle. Many genes are shared between Steinernema and Caenorhabditis, and his lab are now investigating the differences in gene expression between dauers and other larvae in C. elegans.
During his talk Paul highlighted the many resources available for C. elegans researchers, including the CRISPR-based knock-out by knock-in (using the hygromycin resistance cassette) library being created to complement the knockout consortium database of null alleles. He also mentioned the cGAL bipartite expression system that his lab has recently adapted for the worm, and finally his initiatives in micropublication. Micropublication is available through Wormbase and allows concise (one figure) reports to be made and referenced within the scientific community. As he reminds us at the end, the 1953 DNA double helix paper also had just one figure…
Keynote address: Worm Tales by John White
One of the meeting highlights was the keynote address delivered by John White, FRS. John was a graduate student in the lab of Sydney Brenner in the 1970s, and is now Professor Emeritus at the University of Wisconsin-Madison. During his time at the LMB, John co-developed confocal microscopy and published ‘The Mind of a Worm’, and in so doing founded the field of connectomics. His address focused on the historical perspective of modern Caenorhabditis research, from its origin as a brain child of Sydney Brenner to its establishment as a major genetic model organism. (Wormbook has a wonderful memoir describing John White’s experience as part of the first C. elegans research lab that is definitely worth a read.)
In his talk, John explained how Sydney would holiday at the laboratory of marine biology in Woods Hole, contemplating his next scientific adventure (he had already been involved with solving the genetic code, in the process discovering a STOP codon). He was particularly interested in a molecular solution to behaviour, and had the foresight to understand that this would involve a precise understanding of the research organism’s anatomy. However, current model organisms were either too complex (Drosophila has approximately 135,000 neurones) or genetically intractable for this purpose. Sydney came across Bovari’s lineages of the nematode Ascaris, which suggested that these worms were likely to have stereotyped lineages, and also Goldschmidt’s work, which showed Ascaris to have a very simple nervous system. A 1948 paper published in Nature by Dougherty and Calhoun had also suggested the possible significance of nematodes as model organisms. Thus, after some experimentation which showed the nematode nervous system could be visualised with electron microscopy, Sydney settled on the worm as his model. The first strain of Caenorhabditis elegans was isolated from mushrooms in Bristol in England, becoming the ‘canonical’ research strain, and 55 years ago Sydney isolated the first mutant worm.
According to John White, Sydney undertook a very successful PR campaign in the early ’70s to garner support for his new research model. He travelled around America, speaking at many events in order to attract the best and most adventurous postdocs to his laboratory – who also came with funding. People who worked at the LMB with Sydney included many current lab heads and notable researchers: Bob Horwitz, Marty Chalfie, Judith Kimble, Cynthia Kenyon, Barbara Meyer, Jim Preiss, Iva Greenwald, Andy Fire, Julie Ahringer, Benjamin Podbilewitz, John Sulston, Bob Edgar and Ed Hedgecock, amongst others. Graduate students included John White, Jonathan Hodgkin, Mario de Bono, Richard Durbin and Tony Hyman. John explained how Sydney’s approach to supervision could be described as ‘benign neglect’, centring around 10am coffee break discussions that may or may not be related to the lab’s current research. John attributes much of the success of early worm research to this strategy, which forged strong communication and support links between researchers.
Sydney’s first task was to get the genetics of the worm up to the standard of Drosophila, generating mutants and tools such as balancers, and also mapping the C. elegans genome. This work culminated in the publication of a single author paper in 1974. At the same time, the neuroanatomy of the worm was being dissected using electron microscopy. However, Sydney soon moved onto other scientific projects and left C. elegans in the hands of his coworkers. Jonathan Hodgkin took over the construction of the genetic map, and John Sulston decided to help with the effort of White and colleagues to reconstruct the neural network by completing the post-embryonic lineage of the worm. After finding out that those in charge of constructing the embryonic cell lineage were ‘doing it wrong’, Sulston apparently locked himself away for 18 months and produced the accurate map we have today. This work paved the way for the identification of lineage mutants, and the work on cell-cell communication and programmed cell death that was pioneered in the worm. An obituary for John Sulston written by John White can be found here.

John Sulston holding his cell lineage of the nematode C. elegans.
The whole meeting represented a wonderful collection of the continuation of the work initiated by John Sulston, during which he was remembered and missed by the C. elegans community. The historical perspective of John White was a good reminder of how creativity is fostered by the combination of curiosity, tenacity and communication between researchers, values that I believe are central components of the C. elegans research community and make it such a pleasure to be part of.
 (13 votes)
(13 votes)
 (4 votes)
(4 votes) (No Ratings Yet)
(No Ratings Yet)