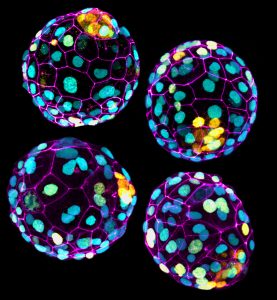
The Sozen laboratory is looking for talented postdoctoral associates with a background in mammalian developmental stem cell biology to join our team. We conduct research in early mouse/human embryogenesis with the goal of understanding the mechanisms that underlie developmental programming in human health and disease.
Our laboratory in the Department of Genetics, Yale School of Medicine, is a part of a highly collaborative, translational developmental biology program that includes stem cell biology, reproductive biology, genetics and computational biology.
The candidate will lead a project to participate in our larger efforts to unravel how the developmental environment impacts on physiologic, metabolic, transcriptomic and epigenomic landscapes during embryo implantation and gastrulation, the time when the maternal-fetal interface is established. The project will integrate cutting-edge technologies such as the in vitro reconstitution of development in 3D with stem cells (Harrison et al. Science 2017, Sozen et al. Nature Cell Bio. 2018, Sozen et al. Dev. Cell 2019), advanced imaging techniques and transcriptomic profiling to map developmental trajectories and cellular function during tissue patterning under environmental challenges.
The postdoctoral candidate will have the opportunity to work in collaboration with a team of world-leading researchers, clinicians, and bioinformaticians, and there will be opportunities to develop additional independent projects with clinical collaborators at Yale University.
In addition to high-quality research facilities, career and professional development training for postdoctoral researchers is provided. Additional information on being a postdoc at Yale University School of Medicine can be found at https://postdocs.yale.edu/
Qualified candidates should have a PhD in Developmental Biology, Stem Cell Biology, Reproductive Biology, Bioengineering or a relevant field. The candidate must have hands-on experience with mammalian embryo culture and/or mammalian stem cell culture. Expertise in 3D cell culture techniques and gene editing methods (e.g. Crispr-Cas9, to engineer stem cell lines with multiple fluorescent reporters) are highly desirable. Experience in transcriptomic profiling techniques (e.g. single-cell RNA-seq) and bioinformatics analyses are desirable but not essential. Please respond with a cover letter that includes a short description of relevant expertise, a CV with publication list, and the names of two references by email to: berna.sozen@yale.edu
On Friday we learned of the death of Lewis Wolpert, a huge figure in developmental biology, a popular science writer and communicator. Here we’ve gathered some memories of Wolpert from the internet – please share your own with a comment below the piece.
Image credit: The British Library
No one who met Lewis could fail to be impressed by his intellect, warmth and humour. Always keen to engage with scientists, regardless of career stage, it was common to see him in animated debates with students and post-docs at conferences. His oratory skills gave him a Thespian aura that could have graced a Shakespearian stage.
Wolpert combined his interest in fundamental problems of development with a parallel career as a science communicator. He enjoyed performing in public, and brooked no compromise in his quest to persuade people that “science is the best way to understand the world”.
The journal Development has asked if I might write about Lewis Wolpert. I’ll cover his science, of course, but Twitter has shown over the last couple of days how kind he was and how many lives he touched. If you have any stories I might include, can you email me? Please re-Tweet!
#LewisWolpert, one of the most influential thinkers in #devbio, has completed his 91 yr developmental journey. As a student in the Tickle & Wolpert lab, I admired how Lewis treated every person, be they an undergraduate or a FRS, as a colleague. He cared about the ideas —status & pic.twitter.com/87laljP8pR
So saddened to hear about the loss of Lewis Wolpert to Covid. An influencer, provocateur and conceptionalist of developmental biology, he will be remembered for defining positional information and gradient models but also for his honesty towards the human fragile condition.
I miss Lewis Wolpert already. I will ever remember the sparkle of warmth and joy in his eyes. He asked me 15 years ago how many cells the mouse embryo needs to survive. We addressed it with @morris_lab and dedicated the paper to him. The number is 4.
I loved Lewis. Wonderful man. An inspiration. Charming, large-hearted, brilliant and wise. I’ll miss the light in his eye when he was trying to start an argument. If there is a God, Lewis will be one atheist she/he’ll definitely want in heaven. Where they’ll be arguing. https://t.co/lf0Zbf7NFM
Yesterday we lost Lewis Wolpert, a giant whose shoulders all #developmental biologists have been blessed to stand in. I had the pleasure to meet him personally when I was a #PhD student @IGCiencia and every year I mention his famous #gastrulation sentence to my students pic.twitter.com/rVUxySo9uN
#LewisWolpert, scientist/thinker extraordinaire, belongs to that breed of scientists who leave an imprint on the field and in the minds and souls of those fortunate enough to interact with them. He had visions and metaphors that still inspire and will inspire many. pic.twitter.com/YLcyQJJt1i
So sad to hear this news. It was Lewis Wolpert’s French Flag model of morphogens that got me into developmental biology. Also when I was a PhD student he sat next to me at a conference breakfast because I was all alone. What an incredible scientist and nice man. https://t.co/D2xftfDUJi
Interacting morphogens produce periodic patterns in developing tissues. Such patterning can be modelled as reaction-diffusion (RD) processes (as originally formulated by Alan Turing), and although these models have been developed and refined over the years, they often tend to oversimplify biological complexity by restricting the number of interacting morphogens. A new paper in Development reports how perturbation analysis can guide multi-morphogen modelling of the striped patterning the roof of the mouse mouth. To hear more about the story, we caught up with first author Andrew Economou and his former supervisor Jeremy Green, Professor of Developmental Biology at King’s College, London.
Andrew (L) and Jeremy (R).
Jeremy, can you give us your scientific biography and the questions your lab is trying to answer?
JG: As an undergraduate I started off studying physics but switched to biochemistry, and I was lucky enough to take the famous Developmental Biology course at Cambridge taught by such luminaries as John Gurdon, Michael Ashburner and Peter Lawrence. That was really what inspired me to follow the subject. For my PhD I got slightly side-tracked into gene expression in yeast, but that was enough to convince me that what I was really interested in was the macro-spatial aspect of biology. I managed to get myself funded to go to a conference where I saw Jim Smith, then a newly-independent investigator, announce his discovery of a mesoderm-inducing factor in Xenopus, and I knew that this was a big deal. Fortunately, Jim took me on as a post-doc and together we did the experiments that showed the inducing factor to be a classic Wolpert French Flag-style morphogen, triggering different cell fates according to dose thresholds.
I then worked with John Gerhart and Ray Keller at UC Berkeley, mostly following up threshold-sharpening with John, but all the while soaking up the morphogenesis culture of the Keller lab. In my own lab (at the Dana-Farber Cancer Institute, part of Harvard Medical School) I then got into cell polarity and Wnt signalling – becoming quite biochemical – but on returning to London and joining King’s College London I re-focused back onto spatial organisation, both with morphogen action and physical morphogenesis.
It has been great harking back to my undergraduate days by approaching this with a physics mind-set, especially working with great collaborators and a mathematically talented postdoc like Andrew.
And Andrew – how did you come to work in Jeremy’s lab and what was the main drive behind your research there?
AE: As a PhD student, I worked in the field of Evolutionary Developmental Biology, looking at the evolution of arthropod segmental identity. Having never had a formal developmental biology training – in my undergraduate I largely specialised in palaeontology – I always felt that there was something missing from the way I thought about embryonic development. After my PhD, I decided that I needed to get a better understanding of the processes that actually shaped the embryo, and it was fortunate that at that time Jeremy was advertising a project for studying the cellular mechanisms underlying the morphogenesis of the mouse face.
When I started with Jeremy, I had no background in any sort of quantitative or computational biology, but it became clear that these approaches were necessary to understanding how the embryo is patterned and shaped. Fortunately, Jeremy was very happy for me to have a go and try to apply computational approaches to my data and see how far we could get. Ultimately, my research became focussed on extracting quantitative information on the different cellular- and molecular-level processes and understanding how they contribute to the distinctive patterning processes seen in the mouse palate epithelium, at the level of both tissue growth and signalling networks.
How has your research been affected by the COVID-19 pandemic?
AE: Like so many other people, I had to put all experiments on hold for the months we were in lockdown. While that obviously held back many parts of my current project (I have moved on from Jeremy’s lab to another postdoc), I have always had some more computational aspect to my work, which I usually keep running in the background. The enforced break from the lab gave me the opportunity to really push on with these parts of my project, so I had plenty of work to keep me occupied! I had also been guilty of allowing a lot of data analysis to build up, so time away from the lab finally gave me a chance to catch up with that. In many ways, this actually helped me understand where I was in my project, and has given me a clearer idea of what my priorities should be since getting back into the lab.
JG: My lab at King’s was shut down completely for a longer time than in some places – partly because we are embedded in a hospital building – so although it has been good for me to catch up on manuscripts, both postdocs and students in the lab are only now (in September) really beginning to pick up momentum again. We were very much in an experimental rather than computational or data analysis phase, so we’re now going to be dependent on supplementary funding from UKRI and King’s to be able to make up for lost time. Fortunately, there are signs that this will be forthcoming, so I’m optimistic.
What have been the main developmental insights – and limitations – of RD modelling before your paper?
AE & JG: Although Turing published his seminal paper almost 70 years ago, we think it’s fair to say that the developmental biology community has only really started to appreciate its importance relatively recently. While we have known for a long time that RD systems can capture biological patterns such as tiger stripes, it has only been in the last 10-20 years that developmental systems have been identified for which RD processes can not only recapitulate the observed patterns, but also for which specific biological molecules have been identified as morphogens.
There has also been a lot of recent theoretical progress, moving beyond the well-established criteria for pattern formation in two-component RD systems, exploring more generally the criteria for the formation of periodic patterns, and looking at how different features of network topology affect the spatial pattern for these larger – more biologically realistic – networks. However, although these larger networks capture more of the complexity of these real biological systems, one thing coming out of these studies was that many networks can produce the same pattern. This is one of the main aspects our paper addresses: how can we use other forms of data, such as inhibiting the different morphogens and following their dynamics during pattern formation, to constrain our understanding of the patterning network?
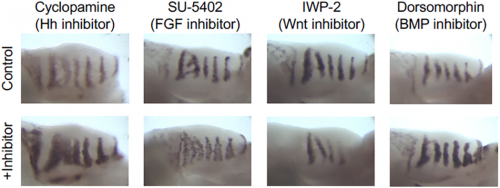
Shh in situ hybridisations on palatal shelf explants cultured in the specified small molecule inhibitor.
Can you give us the key results of the paper in a paragraph?
AE & JG: RD systems are brilliant for describing the self-organising behaviour of morphogens and morphogen-like systems. RD models, from Turing to the very recent past, have been very focused on the minimal two-component system. (Turing’s genius was to strip problems down to the bare minimum and RD is a perfect example of that: two components is all you need.) But biology is messy and evolution isn’t always parsimonious, so in real life there are often more components than you need. As biologists, we want to explain what’s there and see how all these potentially redundant components fit in. Although two-component RD systems have been extensively studied, it is not clear how such simple systems relate to ‘real’ biological networks. We set out to determine how the four signalling pathways Hedgehog, FGF, Wnt and BMP could interact to produce the pattern seen in the palatal rugae – ridges in the roof of the mouse’s mouth. We first established a relatively simple set of experimental observations: how does the pattern respond to the inhibition of each pathway, and what are their spatial patterns of activity? That showed that FGF was working differently in different tissue layers, so we had five rather than four morphogens to deal with. Then, through numerical simulations and mathematical analysis, we established a general framework determining how the response of one component to the inhibition of another is dependent on their position relative to the feedback loops in a minimal RD network, as well as their relative phase. Combining these constraints with temporal information about the sequence of signalling events during the establishment of a rugal stripe, we were able to identify a set of just 154 topologies out of a total set of 39,755 possible networks involving the four pathways, which were consistent with our experimental observations, constraining the sign of all possible interactions in the network.
Biology is messy and evolution isn’t always parsimonious
Rugae stripes appear to be a fairly simple pattern – why do you think it takes five interacting morphogens to make it, when (I guess) two might do the job just as well?
AE & JG: There are three main reasons we can speculate about. One is that the regulatory networks for this system were in effect borrowed from an existing gene regulatory toolkit that happens to include all of these morphogens. There might not be strong selection to get rid of any, so there they are.
The second is that multiple signals might be needed downstream of the stripe formation as such but the system saves time and maybe other kinds of complexity by building them all into the stripe formation. Ruga morphogenesis involves the formation of an epithelial placode and condensation of the underlying mesenchyme, as well as reciprocal signalling between these tissue layers. We suspect that the feedback generated by some of these processes underlies the incorporation of additional pathways into the network after the ruga stripe is initially formed by a subset of pathways.
A third intriguing possibility is that increasing the number of morphogens may increase the robustness of the system. Recent work from other labs has shown that different network topologies vary in their robustness. Therefore, it is not unreasonable to think that initiating a stripe with a system of three morphogens could, for some topologies, be more robust than one made of just two. Although more morphogens provide more targets for perturbations in levels, we were struck by how few of our in silico perturbations changed the stripe spacing and number, so maybe that’s where the robustness needs to be.
How generalisable do you think your approach will be for other developmental problems? And do you have any advice for researchers who wish to increase the number of components in their RD modelling?
AE & JG: Given that our experimental approaches were based on very standard developmental biological techniques – in situ hybridisation and the application of small molecular inhibitors – there is no reason that this approach cannot be applied to other developmental systems patterned by RD processes. Moreover, we think this approach is quite generalisable, even to systems that aren’t obviously periodic. It is known, for example, that Nodal and Lefty proteins constitute a self-organising Turing/RD morphogen pair for left-right patterning in early vertebrates. (The left-right gradient that’s set up is, in effect, a quarter-wavelength of what would be a periodic system if there were enough tissue to encompass it.) So almost any gradient system that involves self-organising feedback might be analysed in much the same way.
One of the most exciting insights from our work, which could be of interest to other researchers, came from looking into the dynamics of stripe formation. Considering the sequence of events which occurred during the formation of the rugae, alongside the feedback we had identified in the system, helped us realise that we can learn more from such a patterning process than just inferring a static depiction of the interactions shown in the network. Different morphogens in a network have different functions: for example, in our system only a subset of the morphogens is actually involved in initiating a new stripe. As the number of components in a network increase, it is important to consider their function in the network.
When doing the research, did you have any particular result or eureka moment that has stuck with you?
AE: The moment that most sticks with me is when I ran my initial set of simulations to look at the effect of inhibiting each component for our set of minimal networks. I had expected that this analysis would pick out a subset of networks but had never really thought about what this would tell us more generally about our system. On seeing the results – that the response of the system to inhibiting different components correlated with the feedback loops they were in – everything made so much more sense and suggested a framework for thinking about much larger networks than the two- and three-component RD systems we were working with.
And what about the flipside: any moments of frustration or despair?
AE: Having run those first simulations and had the initial insight, we then needed a way to actually demonstrate that these results were applicable not just to the two- and three-component systems for which we had run our simulations, but were in theory generalisable to any size of RD system. I had enough of an understanding of the maths to see what needed to be done, but being an experimental biologist actually doing the work was pretty slow progress. Coupled with the fact that I had just started a new postdoc and was therefore having to do a lot of this work in my free time, I sometimes thought we would never get to the end!
What next for you after this paper – I understand you’re now at the Crick?
AE: I have actually been at the Crick for a few years now in Caroline Hill’s lab – this piece of work with Jeremy took a lot longer to wrap up than we initially thought it would! I have been working on early zebrafish development, applying the kinds of computational and quantitative approaches I began to take while in Jeremy’s lab to look at the interactions between signalling pathways during early cell fate decisions in the embryo. I am now looking at options to start my own research group, although in the middle of a pandemic it feels as if this has become even more of a challenge than it normally would be.
Where will this story take the Green lab?
JG: I can see two ways forward with the rugae system. One would be to be more systematic about testing its robustness to perturbation. There’s room for both computational and experimental work to see, for example, whether perturbing two pathways simultaneously does what we predict. Another is to integrate what is really still a fairly minimal system with a transcriptomic picture of what the cells are doing. Have we really captured the key elements of the system, or are there others that we’ve just missed that a transcriptome of the periodicity would reveal?
Finally, let’s move outside the lab – what do you like to do in your spare time in London?
AE: I have 6 and 8 year old sons who have been occupying what spare time I have for the last few years! That said, they are now at an age where we do a lot of fun things together as a family, whether it be going on a bike ride, playing crazy golf or just having a lazy evening watching a film. At times when the work is full on, they certainly force me to switch off!
JG: To my own surprise, I’ve discovered, since working from home, how much I like to get exercise – even a bit of running which I used to hate! My wife and I have been cycling up the Thames in short stages, with a plan to reach the source in a few months, enjoying on the way some beautiful riverside scenery, some lovely little towns, and also a sense of being connected with the world, which is something that rivers give you, I think.
In the latest episode of Genetics Unzipped, the podcast from the Genetics Society, presenter Kat Arney takes a look at the biological changes that underpin ageing, and how we can use this knowledge to live longer, healthier lives.
Kat speaks with Andrew Steele, author of the new book Ageless: The New Science of Getting Older Without Getting Old, to take a deep dive into the processes that underlie ageing and – excitingly – whether we might be able to slow them down. Plus, he explains why the most useful anti-ageing product in your bathroom might be your toothbrush, rather than that fancy moisturiser.
Kat also chats with Raheleh Rahbari, a research fellow at the Wellcome Sanger Institute, who is studying how patterns of DNA damage accumulate in our tissues throughout life. She discusses how our bodies are patchworks of mutation, right from the very start of life, and the impact this has on ageing and diseases like cancer and dementia.
If you enjoy the show, please do rate and review on Apple podcasts and help to spread the word on social media. And you can always send feedback and suggestions for future episodes and guests to podcast@geneticsunzipped.com Follow us on Twitter – @geneticsunzip
The department of Molecular and Cell Biology in the School of Natural Sciences at the University of California, Merced is seeking one new faculty member at the Assistant Professor level (tenure-track) in the area of Molecular Cell Biology. Candidates working on any aspect of molecular and/or cell biology, from the biochemical to organismal level, are encouraged to apply. We seek outstanding scholars who will establish and maintain creative research programs, complement existing strengths in the Molecular and Cell Biology department, align with one or more of the School of Natural Sciences’ areas of strategic focus (Computational & Data Science, Materials, Biomedical Sciences, Sustainability & Environment, and STEM Education), participate in the development of innovative interdisciplinary research projects, and teach effectively at the undergraduate and graduate levels.
Qualifications: A Ph.D. in Molecular Biology, Cell Biology, Biochemistry, Neurobiology, Genetics, Immunology, Computational Biology, or a related field is required. Postdoctoral experience is highly desirable.
This is an immersive laboratory experience designed to provide recent college graduates with additional research experience to make them competitive for advanced careers in biomedical science. This program is focused on providing research experience in modern developmental biology and diverse regenerative medicine labs. Postbaccalaureate researchers will also participate in career development workshops and seminars.
This program is designed to primarily be a research-focused program, where the participants will spend most of their time in the laboratory of a selected mentor. However, it will also provide additional training and opportunities for participants to achieve readiness in three main areas: readiness for research, readiness for academics, and readiness for self-presentation. This will be accomplished through a curriculum that provides educational opportunities in addition to training in the laboratory. Among these will be opportunities to attend tailored workshops, seminars, research forums, and graduate-level classes. A major focus in the second year will be workshops on preparing a Curriculum Vitae and applying to graduate school.
The participant performs research and analysis, as well as technical aspects of studies and experiments, including documentation and preparation of materials.
Individuals accepted into the program will be designated as salaried employees for a period of up to two years and will be eligible for University sponsored benefits, including health insurance.
Works under the supervision of senior personnel on research project/s providing independent complex research support.
Presents results of research activities to peers and supervisors in written and/or verbal presentation or form.
Through active research and analysis gains proficiency in essential aspects of research, which may include some or all of the following: conducting experiments, assays, collection of data, preparation of solutions, tissue culture, animal care and maintenance, and set-up and maintenance of equipment.
Complies with established safety procedures and maintains required documentation on laboratory and specimen conditions.
Attends meetings, educational workshops and seminars to prepare for advanced study.
Performs data entry and maintains data files on research.
Performs other duties as assigned
Required Qualifications
Bachelor’s degree.
Effective verbal, written and interpersonal communication skills with the ability to communicate with laboratory staff and investigators.
Ability to maintain complete and organized records/reports.
The Poché Lab is seeking a highly motivated postdoctoral research associate/fellow with experience in mouse developmental genetics and embryology. This NIH R01-funded position is focused on studying the interplay between nutritional factors, such as vitamins, and genetic and metabolic pathways essential for embryonic development. Special emphasis will be given to neural crest-derived tissues. Our long-term goal is to use this information to devise new therapeutic strategies to modulate the development of neurocristopathies.
Preference will be given to candidates with a strong background in using genetic approaches to study either neural crest or cardiovascular development. This expertise should ideally include next gen sequencing (RNA-seq, ChIP-seq, ATAC-seq, single cell-seq, etc.) data analysis. However, training in these techniques can also be provided.
The Poché lab employs a multi-disciplinary approach utilizing genetic loss- and gain-of-function experiments, fate mapping, gene therapy, molecular biology, and live microscopy. In addition to technical training, all postdocs within the lab routinely receive one-on-one instruction in grant writing and presentation skills.
We are housed in the Department of Molecular Physiology and Biophysics at Baylor College of Medicine (BCM). Located in the Texas Medical Center, the largest medical center on the world, BCM postdocs have a tremendous amount of technical and intellectual resources at their disposal, including the 26 BCM Advanced Technology Core Labs https://www.bcm.edu/research/research-services/atc-core-labs.
In your application, please include a cover letter, current CV, and contact information for three references. Application review will begin immediately and will continue until the position is filled. Please contact Dr. Poché at poche@bcm.edu.
The Poché Lab is seeking a highly motivated postdoctoral research associate/fellow with experience in retinal developmental biology, tissue regeneration, or transcriptome analysis. This NIH R01-funded position is focused on the study of the molecular mechanisms blocking mammalian Müller glial cell (MG)-mediated retinal regeneration. Our long-term goal is to determine whether the mouse retina retains latent regenerative potential, akin to other vertebrate species, and whether we can genetically “awaken” that potential to restore sight.
Special emphasis will be placed on the investigation of MG transcriptional reprogramming to a progenitor-like state. Preference will be given to candidates with a strong background in mouse genetics and in techniques to probe the transcriptome and epigenome. This expertise should ideally include next gen sequencing (RNA-seq, ChIP-seq, ATAC-seq, single cell-seq, etc.) data analysis. However, training in these techniques can also be provided.
The Poché lab employs a multi-disciplinary approach utilizing genetic loss- and gain-of-function experiments, fate mapping, gene therapy, molecular biology, and live retinal confocal microscopy. In addition to technical training, all postdocs within the lab routinely receive one-on-one instruction in grant writing and presentation skills.
We are housed in the Department of Molecular Physiology and Biophysics at Baylor College of Medicine (BCM). Located in the Texas Medical Center, the largest medical center on the world, BCM postdocs have a tremendous amount of technical and intellectual resources at their disposal, including the 26 BCM Advanced Technology Core Labs https://www.bcm.edu/research/research-services/atc-core-labs.
In your application, please include a cover letter, current CV, and contact information for three references. Application review will begin immediately and will continue until the position is filled. Please contact Dr. Poché at poche@bcm.edu.
The next webinar in our Development presents… series will be a little different: rather than being chaired by a Development editor, the preLights team will be in control. preLights is the preprint highlights service run by the biological community and supported by The Company of Biologists, and in February they celebrate their third birthday.
We brought together three preLighters with interests in developmental biology – Sundar Naganathan, Irepan Salvador-Martinez and Grace Lim – who have each invited authors of recent exciting preprints to give talks. The preLighters will chair the talks and the Q&As, and we’ll also hear from outgoing preLights Community Manager Mate Palfy about three years in the life of a preprint-focused community. We hope to see you there!
Wednesday 10 February 2021 – 13:00 GMT
MichèleRomanos (from Bertrand Benazeraf’s lab at the Centre de Biologie Integrative in Toulouse)
‘Cell-to-cell heterogeneity in Sox2 and Brachyury expression ratios guides progenitor destiny by controlling their motility.’
The webinar will be held in Remo, our browser-based conferencing platform – after the talks you’ll have the chance to meet the speakers and other participants at virtual conference tables. If you can’t make it on the day, talks will be available to watch for a couple of weeks after the event (look out for details on the Node).
For more information about what to expect in Remo, go to
The employment is for 4 years, as research assistant is for 1 year and as PhD fellow for the following 3 years, and is scheduled to start in July or upon agreement with the chosen candidate.
The Novo Nordisk Foundation Center for Stem Cell Biology (DanStem) at Faculty of Health & Medical Sciences at the University of Copenhagen is looking for a Research assistant subsequent appointed as PhD fellow to join the Żylicz group starting July 2021 or upon agreement with the chosen candidate.
The position as Research assistant is for 1 year. The position as PhD fellow is for 3 years.
This position is available in the group of Jan Żylicz at DanStem. The team studies fundamental mechanisms of early mouse development and stem cell biology. The Żylicz group wants to understand how metabolic and epigenetic mechanisms cooperate to regulate transcription during early development. In particular the team is interested in how metabolism regulates histone modifiers, and how these in turn affect lineage choice and embryo growth at around the time of implantation. To achieve this, the group utilizes both in vivo mouse models as well as in vitro stem cell culture systems and state-of the-art ultrasensitive transcriptomic, epigenomic and metabolomic techniques. This research project will employ a multi-disciplinary approach to understand how first lineage choices are influenced by metabolic and chromatin states. Starting date
for this position is the 1 July 2021, or upon agreement with the chosen candidate.
Job description:
We are seeking a highly motivated and ambitious pre-doctoral candidate with a strong educational background in either stem cell and developmental biology and/or metabolomic research and/or bioinformatics. The candidate will investigate the molecular mechanisms controlling first lineage choice of early development using both in vivo and in vitro stem cell models. By employing genomic engineering methods the candidate will go beyond description of epigenetic and metabolic states and uncover novel functional regulatory mechanisms at the interplay of epigenetics and metabolism.
About DanStem:
The Novo Nordisk Foundation Center for Stem Cell Biology – DanStem – addresses basic research questions in stem cell and developmental biology and translates results from basic research into new strategies and targets for the development of new therapies for diabetes and cancer. DanStem was established as a result of a series of international recruitments coupled with internationally recognized research groups focused on insulin producing beta cells and cancer research already located at the University of Copenhagen.
Qualifications:
Candidates must hold a master’s degree in biotechnology, bio-informatics, biology or similar, and possess a strong background in developmental and stem cell biology and/or metabolomics and/or bioinformatics.
Previous practical experience in bioinformatics, analysis of epigenomic data and/or software programming is considered of great advantage.
Previous experience using rodents as a research model, stem cell culture, embryogenesis and/or next-generation sequencing are considered an advantage.
Practical project experience in basic lab techniques is considered beneficial.
Good English communication skills, both oral and written, are prerequisite for the successful candidate.
A solution-oriented, organizational and positive mindset is required. The ability to work in a highly diverse and collaborative environment both independently and as part of the team is essential.
Terms of salary, work, and employment:
The employment is for 4 years, as research assistant is for 1 year and as PhD fellow for the following 3 years, and is scheduled to start in July or upon agreement with the chosen candidate. The employment as a PhD student is conditioned upon a positive assessment of the candidate ́s research performance and enrolment in the Graduate School at the Faculty of Health and Medical Sciences.
Salary, pension and terms of employment are in accordance with the provisions of the collective agreement between the Danish Government and AC (the Danish Confederation of Professional Associations). In addition to the basic salary a monthly contribution to a pension fund is added (17.1% of the salary).
The PhD study must be completed in accordance with the ministerial orders from the Ministry of Education on the PhD degree and the University ́s rules on achieving the degree.
The place of work is at DanStem, University of Copenhagen, Blegdamsvej 3B, Copenhagen.
Questions:
For further information please contact Associate Professor Jan Żylicz, jan.zylicz@sund.ku.dk.
The application must include:
Motivation letter
Curriculum vitae incl. education, experience, previous employments, language skills and other relevant skills
List of two references (full address, incl. email and phone number)
Copy of diplomas/degree certificate(s)
How to apply
The application, in English, must be submitted electronically by clicking APPLY below.
Application deadline: 21 February 2021
Only applications received in time and consisting of the above listed documents will be considered.
Applications and/or any material received after deadline will not be taken into consideration.
The University of Copenhagen wishes to reflect the diversity of society and welcomes applications from all qualified candidates regardless of personal background.
The application will be assessed according to the Ministerial Order no. 284 of 25 April 2008 on the Appointment of Academic Staff at Universities.
Assessment procedure
After the expiry of the deadline for applications, the authorized recruitment manager selects applicants for assessment on the advice of the Appointments Committee. All applicants are then immediately notified whether their application has been passed for assessment by an expert assessment committee. Selected applicants are notified of the composition of the committee and each applicant has the opportunity to comment on the part of the assessment that relates to the applicant him/herself. You can read about the recruitment process at http://employment.ku.dk
Københavns Universitet giver sine knap 10.000 medarbejdere muligheder for at udnytte deres talent fuldt ud i et ambitiøst, uformelt miljø. Vi sikrer traditionsrige og moderne rammer om uddannelser og fri forskning på højt internationalt niveau. Vi søger svar og løsninger på fælles problemer og gør ny viden tilgængelig og nyttig for andre.
Info
Application deadline: 21-02-2021
Date of employment: 01-07-2021
Working hours: Full time
Department / Place: The Novo Nordisk Foundation Center for Stem Cell Biology
 The Sozen laboratory is looking for talented postdoctoral associates with a background in mammalian developmental stem cell biology to join our team. We conduct research in early mouse/human embryogenesis with the goal of understanding the mechanisms that underlie developmental programming in human health and disease.
The Sozen laboratory is looking for talented postdoctoral associates with a background in mammalian developmental stem cell biology to join our team. We conduct research in early mouse/human embryogenesis with the goal of understanding the mechanisms that underlie developmental programming in human health and disease. (3 votes)
(3 votes)

 (No Ratings Yet)
(No Ratings Yet)
