The correct patterning of embryonic tissues is essential for normal development. Aberrant patterning can lead to developmental abnormalities and pathogenic defects. Therefore, studying developmental patterning is important to better understand disease. The zebrafish embryo is a fantastic model for studying patterning during development owing to its optical clarity, small size and large clutch number. When coupled with dynamic transgenic lines, the picture of what occurs in the cell during these processes is starting to emerge.
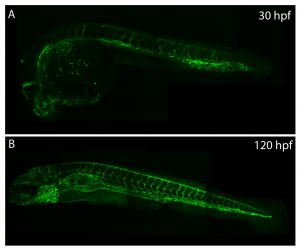
The vasculature of zebrafish expands throughout development, providing developing tissues with oxygen and nutrients.
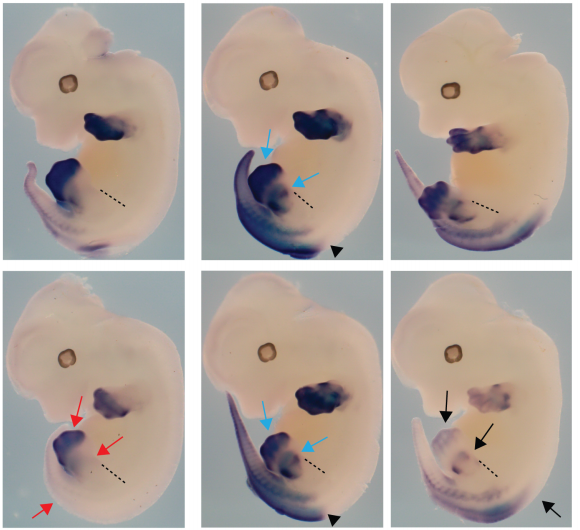
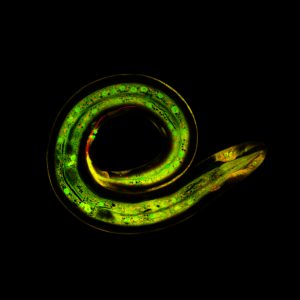
Understanding how the ubiquitous second messenger, the calcium ion, regulates cellular physiology during development has become an important question in biology. This is where my story begins. I arrived in Sheffield after obtaining a PhD position, eager for what lay ahead. The project was exciting; using new transgenic lines and cutting-edge microscopy to study the function of a poorly understood gene, tmem33, which we hypothesised would regulate calcium signalling within the developing vasculature. The preliminary morpholino knockdown data generated in my host lab before I started my PhD hinted at a vast cache of riches waiting to be uncovered. This data suggested a role for tmem33 in both vascular and kidney development. I was given some new toys to play with, a light sheet microscope and an endothelial-specific calcium reporter line. My first experiment in the lab was to analyse the mutant zebrafish line generated by my supervisor around which my PhD was supposed to focus. It was, of course, completely normal. No vascular defects here. It looked like my project was dead in the water before it had begun. My next few experiments yielded additional dead ends – analysing 34 TRP channels by in situ hybridisation, looking for specific vascular enrichment (spoiler alert – there wasn’t any).
Thankfully, science moves very quickly. My original project went from dead, to very dead as several papers at the time heavily criticised morpholino-based approaches (Kok et al, 2014; Schulte-Merker and Stainier, 2014; Stainier 2017; Robu, 2007) to back alive within the space of about six months. A single paper (Rossi et al, 2015) brought hopes for a revived project. In a series of elegant experiments, the authors described a situation where knockdown of the egfl7 gene by morpholino induced a robust vascular phenotype, but when this gene was mutated using genome editing the phenotype was absent. Interestingly, the authors showed that the egfl7 mutant displayed nonsense mediated decay of egfl7 transcripts and that when egfl7 morpholinos were injected into egfl7 mutants, the mutants were protected against the effects of the morpholino. The synthesis of these data was that there existed genetic compensation in the mutants but not in the morphants.
Finally, the stroke of luck I needed, our tmem33 mutants also showed nonsense mediated decay, so I set about injecting tmem33 morpholinos into our tmem33 mutants and I found strikingly similar results. This suggested that the reason the tmem33 mutants displayed no phenotype was because they displayed a kind of genetic protection, likely via a genetic compensation mechanism. I now had the beginnings of a successful project, a year in to my PhD. Rossi et. al used a new technology to address their issues, CRISPR interference (CRISPRi) – a modified version of the CRISPR/Cas9 system using an inactivated form of Cas9 (dCas9). The authors were able to reproduce the same phenotype they observed via morpholino knockdown using CRISPRi. I applied CRISPRi to knock down tmem33 and was able to reproduce our morpholino knockdown data of tmem33.
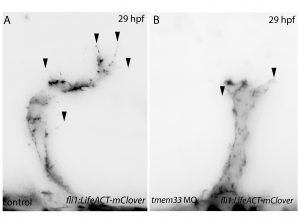
Filopodia are essential for cellular migration. Tmem33 morphants display reduced filopodia and delayed vascular migration.
Next, inspired by the conditional CRISPR approaches described in Ablain et al (2015), I sought to conditionally knock down tmem33 in endothelial cells by driving dCas9 expression under the control of the endothelial-specific fli1a promoter. Preliminary experiments using transient conditional knockdown were promising – only endothelial cells showed a phenotype! The next step was to generate a transgenic line which stably expressed dCas9 in endothelial cells.
Throughout this process, I had been studying tmem33 function with regards to endothelial cell physiology during angiogenesis. Since it was known that tmem33 functioned within the endoplasmic reticulum, using a calcium reporter line I began to test whether tmem33 knockdown altered endothelial calcium signalling. I found that tmem33 knockdown reduced observable endothelial calcium oscillations, which coincided with reduced endothelial cell migration. This was validated by both morpholinos and CRISPRi. Furthermore, I began using both knockdown approaches to position the tmem33 within the hierarchy of developmental angiogenic signalling. I found that tmem33 functions downstream of VEGF signalling but upstream of Notch and ERK signalling, identifying an essential function for calcium oscillations in mediating the response to Vascular Endothelial Growth Factor (VEGF) and inducing downstream signalling pathways essential during angiogenesis.
Calcium signalling during endothelial development.
Throughout my PhD, I’ve learned how the zebrafish can be a powerful tool for understanding basic biology and how a PhD is not a linear endeavour. You have to follow the research (and the results of others) and see where it takes you. I’ve also found that phenotypes (or rather, the lack of them) can be hard to interpret. If you think about it, nothing would be alive today if there weren’t contingency plans built into the genome. Unexpected negative results (I’m looking at you again, non-phenotypic mutants) are not worthless. In fact, they’re probably more interesting.
References
Kok, Fatma O., et al. “Reverse genetic screening reveals poor correlation between morpholino-induced and mutant phenotypes in zebrafish.” Developmental cell 32.1 (2015): 97-108.
Robu, Mara E., et al. “p53 activation by knockdown technologies.” PLoS genetics 3.5 (2007): e78.
Rossi, Andrea, et al. “Genetic compensation induced by deleterious mutations but not gene knockdowns.” Nature 524.7564 (2015): 230.
Savage, Aaron M., et al. “tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis.” Nature communications 10.1 (2019): 732.
Schulte-Merker, Stefan, and Didier YR Stainier. “Out with the old, in with the new: reassessing morpholino knockdowns in light of genome editing technology.” Development 141.16 (2014): 3103-3104.
Stainier, Didier YR, et al. “Guidelines for morpholino use in zebrafish.” PLoS genetics 13.10 (2017): e1007000.
A postdoctoral position is available immediately for a highly-motivated individual interested in understanding the molecular mechanisms of brain development. The individual will join Dr. Martin Riccomagno’s laboratory at the University of California, Riverside (http://www.riccomagnolab.org). Our laboratory uses a combination of molecular biology, histology, tissue culture, mouse transgenesis, and cutting-edge imaging approaches to investigate the developmental mechanisms that regulate neural circuit formation.
The successful applicant will be involved in NIH-funded projects in one of the following research areas:
1) Regulation of neural migration by adhesive cues
2) Developmental profiling of caspase-dependent refinement
The salary for this position is commensurate with experience and determined by NIH guidelines.
Position requirements:
A PhD or MD-PhD degree in a biological science
A solid background in neuroscience or developmental biology
Previous experience working with genetic mouse models
Proficiency in cellular and molecular biology
Excellent communication skills
A demonstrated ability to work independently and learn new techniques
The scientific environment at UC Riverside is highly collaborative and multidisciplinary. One of the most significant resources available is the quantity and quality of valuable scientific interaction and support from fellow cell biologists and neuroscientists both within and beyond the Department of Molecular, Cell and Systems Biology (MCSB). UCR is also home to the Center for Glial-Neuronal Interactions (CGNI). CGNI provides a forum for scientific interactions among the members of the UCR neuroscience community, and organizes an annual research symposium that attracts neuroscientists from throughout southern California, and speakers from around the world.
Interested applicants should apply by sending a cover letter, CV, and contact information for three references to: martinmr@ucr.edu
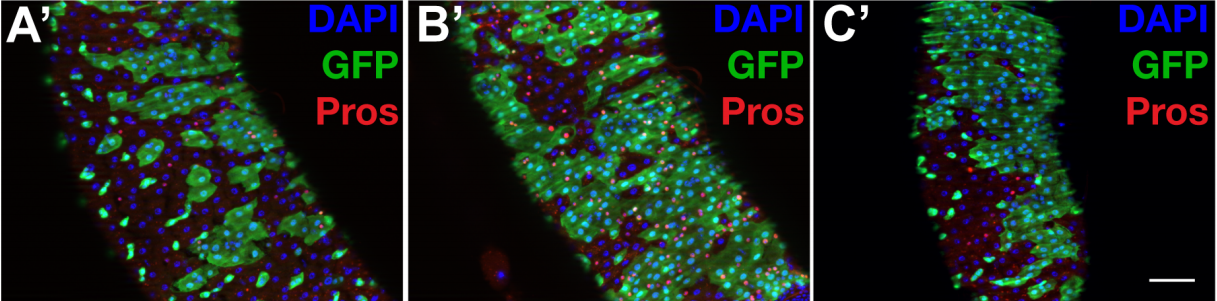
With air pollution on the rise, our respiratory system is continually abused by a barrage of harmful substances that we breathe in with each inhalation. Fortunately, we are equipped with highly specialised ciliated cells, the multiciliated cells (MCCs), which differentiate hundreds of motile cilia on their apical surface1,2. These cilia beat rhythmically to drive mucus that entraps air borne toxins and pathogens out of the airways – a process called mucociliary clearance in clinical circles. The importance of mucociliary clearance in respiratory physiology is perhaps best exemplified by patients afflicted with diseases, genetic as well as acquired, in which the cilia on MCCs do not function properly. In such instances, individuals suffer from recurrent lung infections that often lead to irreparable damage called bronchiectasis3. Given their importance, it is understandable why the MCCs have lately attracted a lot of attention. MCCs are also present in organs such as the central nervous system (within the brain ventricles and spinal canal), where they promote circulation of cerebrospinal fluid (CSF), and within the reproductive system where they are required for mixing of reproductive fluids and germ cell transportation. Pathological consequences of defective MCCs in these organs include hydrocephalus (distention of the brain ventricles due to improper CSF flow) and infertility, respectively1,2.
A particularly intriguing aspect of the MCC differentiation program is the ability of their precursors to support an explosive production of basal bodies (specialised centrioles), which are required to nucleate the assembly of multiple cilia. While most cells duplicate centrioles only once during the cell cycle, how post-mitotic MCC precursors can generate hundreds of these organelles has intrigued cell biologists for decades. However, it is only over the past few years that we have made substantial progress in identifying some of the key genetic determinants and important cell biological pathways that govern the differentiation of the MCCs. Notably, in a seminal piece of work published in 2012, Kinter and colleagues identified a small coiled-coil containing nuclear protein Multicilin (aka Mcidas), related to the celebrated DNA replication factor Geminin, as necessary and sufficient for MCC development4. In their study, which mainly utilized Xenopus embryos that differentiate MCCs on the epidermis, they could show that morpholino-mediated inhibition of Mci function completely abolished MCCs, while its overexpression led to the production of ectopic MCCs at the expense of other epidermal cell-types. Since Mci lacks a DNA binding domain, when a subsequent paper by the same group showed that the protein functions in association with the E2f family of cell-cycle transcription factors to activate MCC-specific gene expression (genes for production of multiple basal bodies and cilia)5, our understanding of MCC development achieved a certain degree of completeness. More importantly, mutations in MCI were concomitantly identified in patients with the respiratory disorder reduced generation of multiple motile cilia, underscoring the importance of the MCC developmental pathway in the etiology of severe airway disease6.
It was soon realised though, that there is more to MCCs than Mci. We and others found that another Mci-like protein, Gmnc (aka Gemc1), has identical effects on MCC development. Zebrafish, Xenopus and mice deficient in Gmnc function completely lack MCCs from various tissues in which these cells normally differentiate7,8,9. Moreover, overexpression of Gmnc can drive the production of supernumerary MCCs. In addition, these studies also showed that Gmnc functions upstream of Mci, since Mci is not induced in the absence of Gmnc, but Mci is incapable of inducing Gmnc expression. Despite all of this information, one outstanding question that has lingered in the field is how the activities of two highly related proteins can have near identical effects on the MCC developmental program –both proteins being necessary as well as sufficient for MCC development. We reasoned that evaluating the function of Mci with a stable genetic mutant in the mouse would be the best way forward, as the phenotypes that have been described for the Gmnc mutant mice will allow the appropriate comparisons to be made, in the context of a mammal. Surprisingly, in contrast to the current notion of Mci function, which posits its requirement in MCC specification as well as differentiation, we now report in Development that mice lacking Mci function can generate MCC precursor cells in normal numbers10. These precursors can express the full suite of genes that have been linked to the transcriptional regulation of ciliogenesis. However, they are completely unable to generate any basal bodies for multiciliation, and instead differentiate a single motile-like cilium.
A multiciliated cell from the human airway. Cilia are stained with antibodies to acetylated tubulin (red) and the nucleus is labelled with DAPI (blue).
Moreover, we could show that while Gmnc can activate genes that regulate motile ciliogenesis, Mci preferentially activates genes that promote the biogenesis of multiple basal bodies. Finally, we were also able to demonstrate that the difference in the transcriptional activities of the two proteins possibly lies in the differences in their ability to associate with E2f4 versus E2f5, and thereby target the regulation of different sets of MCC-specific differentiation genes. In sum, our study has clarified that Gmnc functions at the top of the transcriptional hierarchy for making MCCs: it induces MCC precursors and also activates the expression of Mci. Subsequently, Mci programs these cells to generate multiple basal bodies and complete the process of multiciliation10.
So what’s next for Mci? While we do need a much better understanding of the molecular basis of Mci-mediated transcriptional regulation, the future looks even brighter for the protein to enable us to tackle a more profound and unsolved issue in MCC development: What is the precise cellular pathway that allows the MCCs to generate multiple basal bodies? Current view posits that there are two ways by which this is achieved. First, in the mother centriole-dependant pathway, the mother centriole is thought to produce a small number of centrioles (this is the pathway utilised for centriole duplication during regular cell cycle). However, it is via a unique MCC-specific alternative pathway, the deuterosome-dependant pathway, that more than 90% of the basal bodies are produced. Although deuterosomes, electron dense ring-shaped structures that function in centriole biogenesis in MCCs, were originally believed to arise de novo, more recent evidence has suggested that they are produced in close association with the daughter centriole11,12,13. Research into the mechanism by which MCCs make multiple basal bodies is in such a state of flux, that accumulating new data are again poised to revise our present view of the contributions the two pathways highlighted above. Three papers, all in preprint, make the tantalising proposition that in fact the centrosomal centrioles are completely dispensable for basal body generation in the MCCs14,15,16!
Sudipto Roy is a Senior Principal Investigator with the Institute of Molecular and Cell Biology, A*STAR, Singapore.
In the absence of the mother and the daughter centrioles, MCC precursors can generate deuterosomes quite normally, likely from the pericentriolar material (PCM) associated with the microtubule organizing centre (MTOC), and these deuterosomes produce the normal complement of basal bodies. Given that Mci mutant MCCs are totally incapable of generating multiple basal bodies, we can look forward to obtaining useful insights into this enigmatic process through more detailed investigations into the basal body generation defects of the MCC precursors in these mice. In the end, all of this information will add significant value for appreciating the underlying mechanistic basis of the pathological consequences associated with ciliary disorders that affect the formation of the MCCs and the motility of their cilia.
References:
BROOKS, ERIC R. & WALLINGFORD, JOHN B. 2014. Multiciliated Cells. Current Biology 24, R973-R982.
ZHOU, F. & ROY, S. 2015. SnapShot: Motile Cilia. Cell 162, 224-224.e1.
BUSTAMANTE-MARIN, X. M. & OSTROWSKI, L. E. 2017. Cilia and Mucociliary Clearance. Cold Spring Harb. Perspect. Biol. 9, a028241.
STUBBS, J. L., VLADAR, E. K., AXELROD, J. D. & KINTNER, C. 2012. Multicilin promotes centriole assembly and ciliogenesis during multiciliate cell differentiation. Cell Biol. 14, 140-7.
MA, L., QUIGLEY, I., OMRAN, H. & KINTNER, C. 2014. Multicilin drives centriole biogenesis via E2f proteins. Genes Dev. 28, 1461-71.
BOON, M., WALLMEIER, J., MA, L., LOGES, N. T., JASPERS, M., OLBRICH, H., DOUGHERTY, G. W., RAIDT, J., WERNER, C., AMIRAV, I., HEVRONI, A., ABITBUL, R., AVITAL, A., SOFERMAN, R., WESSELS, M., O’CALLAGHAN, C., CHUNG, E. M., RUTMAN, A., HIRST, R. A., MOYA, E., MITCHISON, H. M., VAN DAELE, S., DE BOECK, K., JORISSEN, M., KINTNER, C., CUPPENS, H. & OMRAN, H. 2014. MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia. Commun. 5, 4418.
ARBI, M., PEFANI, D.-E., KYROUSI, C., LALIOTI, M.-E., KALOGEROPOULOU, A., PAPANASTASIOU, A. D., TARAVIRAS, S. & LYGEROU, Z. 2016. GemC1 controls multiciliogenesis in the airway epithelium. EMBO Rep. 17, 400-13.
TERRÉ, B., PIERGIOVANNI, G., SEGURA-BAYONA, S., GIL-GÓMEZ, G., YOUSSEF, S. A., ATTOLINI, C. S.-O., WILSCH-BRÄUNINGER, M., JUNG, C., ROJAS, A. M., MARJANOVIĆ, M., KNOBEL, P. A., PALENZUELA, L., LÓPEZ-ROVIRA, T., FORROW, S., HUTTNER, W. B., VALVERDE, M. A., DE BRUIN, A., COSTANZO, V. & STRACKER, T. H. 2016. GEMC1 is a critical regulator of multiciliated cell differentiation. EMBO J. 35, 942-60.
ZHOU, F., NARASIMHAN, V., SHBOUL, M., CHONG, Y. L., REVERSADE, B. & ROY, S. 2015. Gmnc is a master regulator of the multiciliated cell differentiation program. Biol. 25, 3267-73.
LU, H., ANUJAN, P., ZHOU, F., ZHANG, Y., CHONG, Y. L., BINGLE, C. D., & ROY, S. 2019. Mcidas mutant mice reveal a two-step process for the specification and differentiation of multiciliated cells in mammals. Development 146: dev172643 doi: 10.1242/dev.172643
SOROKIN, S. P. 1968. Reconstructions of centriole formation and ciliogenesis in mammalian lungs. Cell Sci. 3, 207-30.
ZHAO, H., ZHU, L., ZHU, Y., CAO, J., LI, S., HUANG, Q., XU, T., HUANG, X., YAN, X. & ZHU, X. 2013. The Cep63 paralogue Deup1 enables massive de novo centriole biogenesis for vertebrate multiciliogenesis. Cell. Biol. 15, 1434-44.
AL JORD, A., LEMAITRE, A. I., DELGEHYR, N., FAUCOURT, M., SPASSKY, N. & MEUNIER, A. 2014. Centriole amplification by mother and daughter centrioles differs in multiciliated cells. Nature 516, 104-7.
MERCEY, O., AL JORD, A., ROSTAING, P., MAHUZIER, A., FORTOUL, A., BOUDJEMA, A.-R., FAUCOURT, M., SPASSKY, N. & MEUNIER, A. 2018. Dynamics of centriole amplification in centrosome-depleted brain multiciliated progenitors. bioRxiv, 503730.
NANJUNDAPPA, R., KONG, D., SHIM, K., STEARNS, T., BRODY, S., LONCAREK, J. & MAHJOUB, M. 2018. Regulation of cilia abundance in multiciliated cells. bioRxiv, 478297.
ZHAO, H., CHEN, Q., HUANG, Q., YAN, X. & ZHU, X. 2018. Mother centrioles are dispensable for deuterosome formation and function during basal body amplification. bioRxiv, 373662.
Welcome to our monthly trawl for developmental biology (and related) preprints.
This month we found three hydra preprints, lots of developmental mechanics, a typically hearty serving of single cell transcriptomic analyses and a survey of the life of PIs.
The preprints were hosted on bioRxiv, PeerJ, andarXiv. Let us know if we missed anything, and use these links to get to the section you want:
Microglia actively remodels adult hippocampal neurogenesis through the phagocytosis secretome
Irune Diaz-Aparicio, Iñaki Paris, Virginia Sierra-Torre, Ainhoa Plaza-Zabala, Noelia Rodríguez-Iglesias, Mar Márquez-Ropero, Sol Beccari, Oihane Abiega, Elena Alberdi, Carlos Matute, Irantzu Bernales, Angela Schulz, Lilla Otrokocsi, Beata Sperlagh, Kaisa E. Happonen, Greg Lemke, Mirjana Maletic-Savatic, Jorge Valero, Amanda Sierra
Neuronal programming by microbiota enables environmental regulation of intestinal motility
Yuuki Obata, Stefan Boeing, Álvaro Castaño, Ana Carina Bon-Frauches, Mercedes Gomez de Agüero, Werend Boesmans, Bahtiyar Yilmaz, Rita Lopes, Almaz Huseynova, Muralidhara Rao Maradana, Pieter Vanden Berghe, Andrew J. Murray, Brigitta Stockinger, Andrew J. Macpherson, Vassilis Pachnis
Human cortical organoids expose a differential function of GSK3 on direct and indirect neurogenesis
Alejandro López-Tobón, Carlo Emanuele Villa, Cristina Cheroni, Sebastiano Trattaro, Nicolò Caporale, Paola Conforti, Raffaele Iennaco, Maria Lachgar, Marco Tullio Rigoli, Berta Marcó de la Cruz, Pietro Lo Riso, Erika Tenderini, Flavia Troglio, Marco de Simone, Isabel Liste-Noya, Stefano Piccolo, Giuseppe Macino, Massimiliano Pagani, Elena Cattaneo, Giuseppe Testa
Pluripotency factors regulate the onset of Hox cluster activation in the early embryo
Elena Lopez-Jimenez, Julio Sainz de Aja, Claudio Badia-Careaga, Antonio Barral, Isabel Rollan, Raquel Rouco, Elisa Santos, María Tiana, Jesus Victorino, Hector Sanchez-Iranzo, Rafael D Acemel, Carlos Torroja, Javier Adan, Eduardo Andres-Leon, Jose Luis Gomez-Skarmeta, Giovanna Giovinazzo, Fatima Sanchez-Cabo, Miguel Manzanares
Embryonic stem cells from Rhodes, et al.’s preprint
Cohesin disrupts polycomb-dependent chromosome interactions
JDP Rhodes, A Feldmann, B Hernández-Rodríguez, N Díaz, JM Brown, NA Fursova, NP Blackledge, P Prathapan, P Dobrinic, M Huseyin, A Szczurek, K Kruse, KA Nasmyth, VJ Buckle, JM Vaquerizas, RJ Klose
Whole-genome and RNA sequencing reveal variation and transcriptomic coordination in the developing human prefrontal cortex
Donna M. Werling, Sirisha Pochareddy, Jinmyung Choi, Joon-Yong An, Brooke Sheppard, Minshi Peng, Zhen Li, Claudia Dastmalchi, Gabriel Santpere, Andre M. M. Sousa, Andrew T. N. Tebbenkamp, Navjot Kaur, Forrest O. Gulden, Michael S. Breen, Lindsay Liang, Michael C. Gilson, Xuefang Zhao, Shan Dong, Lambertus Klei, A. Ercument Cicek, Joseph D. Buxbaum, Homa Adle-Biassette, Jean-Leon Thomas, Kimberly A. Aldinger, Diana R. O’Day, Ian A. Glass, Noah A. Zaitlen, Michael E. Talkowski, Kathryn Roeder, Matthew W. State, Bernie Devlin, Stephan J. Sanders, Nenad Sestan
Human cortical neural stem cells generate regional organizer states in vitro before committing to excitatory neuronal fates
Nicola Micali, Suel-Kee Kim, Marcelo Diaz-Bustamante, Genevieve Stein-O’Brien, Seungmae Seo, Joo-Heon Shin, Brian G. Rash, Shaojie Ma, Nicolas A. Olivares, Jon Arellano, Kristen R. Maynard, Elana J. Fertig, Alan J. Cross, Roland Burli, Nicholas J. Brandon, Daniel R. Weinberger, Joshua G. Chenoweth, Daniel J. Hoeppner, Nenad Sestan, Pasko Rakic, Carlo Colantuoni, Ronald D. McKay
Identification of a human adult cardiac stem cell population with neural crest origin
Anna Höving, Madlen Merten, Kazuko Elena Schmidt, Isabel Faust, Lucia Mercedes Ruiz-Perera, Henning Hachmeister, Sebastian-Patrick Sommer, Buntaro Fujita, Thomas Pühler, Thomas Huser, Johannes Greiner, Barbara Kaltschmidt, Jan Gummert, Cornelius Knabbe, Christian Kaltschmidt
The Gag Protein PEG10 Binds to RNA and Regulates Trophoblast Stem Cell Lineage Specification
Mona Abed, Erik Verschueren, Hanna Budayeva, Peter Liu, Donald S. Kirkpatrick, Rohit Reja, Sarah K. Kummerfeld, Joshua D. Webster, Sarah Gierke, Mike Reichelt, Keith R. Anderson, Robert J Newman, Merone Roose-Girma, Zora Modrusan, Hazal Pektas, Emin Maltepe, Kim Newton, Vishva M. Dixit
A smooth muscle-like niche facilitates lung epithelial regeneration
Alena Moiseenko, Ana Ivonne Vazquez-Armendariz, Xuran Chu, Stefan Guenther, Kevin Lebrigand, Vahid Kheirollahi, Susanne Herold, Thomas Braun, Bernard Mari, Stijn De Langhe, Chengshui Chen, Xiaokun Li, Werner Seeger, Jin-San Zhang, Saverio Bellusci, Elie El Agha
Human liver organoids; a patient-derived primary model for HBV Infection and Related Hepatocellular Carcinoma
Elisa De Crignis, Fabrizia Carofiglio, Panagiotis Moulos, Monique M.A. Verstegen, Shahla Romal, Mir Mubashir Khalid, Farzin Pourfarzad, Christina Koutsothanassis, Helmuth Gehart, Tsung Wai Kan, Robert-Jan Palstra, Charles Boucher, Jan M.N. Ijzermans, Meritxell Huch, Sylvia F. Boj, Robert Vries, Hans Clevers, Luc van der Laan, Pantelis Hatzis, Tokameh Mahmoudi
Glioblastomas derived from genetically modified pluripotent stem cells recapitulate pathobiology
Tomoyuki Koga, Jorge A Benitez, Isaac A Chaim, Sebastian Markmiller, Alison D Parisian, Kristen M Turner, Florian M Hessenauer, Matteo D’Antonio, Nam-phuong D Nguyen, Shahram Saberi, Jianhui Ma, Shunichiro Miki, Antonia D Boyer, John Ravits, Kelly A Frazer, Vineet Bafna, Clark C Chen, Paul S Mischel, Gene W Yeo, Frank B Furnari
Molecular mechanisms of Evening Complex activity in Arabidopsis
Catarina S. Silva, Aditya Nayak, Xuelei Lai, Veronique Hugouvieux, Jae-Hoon Jung, Agnès Jourdain, Irene López-Vidriero, Jose Manuel Franco-Zorrilla, François Parcy, Kishore Panigrahi, Philip A. Wigge, Max Nanao, Chloe Zubieta
The quail as an avian model system: its genome provides insights into social behaviour, seasonal biology and infectious disease response
Katrina Morris, Matthew M Hindle, Simon Boitard, David W Burt, Angela F Danner, Lel Eory, Heather L Forrest, David Gourichon, Jerome Gros, LaDeana Hillier, Thierry Jaffredo, Hanane Khoury, Rusty Lansford, Christine Leterrier, Andrew Loudon, Andrew S Mason, Simone L Meddle, Francis Minvielle, Patrick Minx, Frederique Pitel, J Patrick Seiler, Tsuyoshi Shimmura, Chad Tomlinson, Alain Vignal, Robert G Webster, Takashi Yoshimura, Wesley C Warren, Jacqueline Smith
A draft genome sequence of the miniature parasitoid wasp, Megaphragma amalphitanum
Artem V. Nedoluzhko, Fedor S. Sharko, Brandon M. Lê, Svetlana V. Tsygankova, Eugenia S. Boulygina, Sergey M. Rastorguev, Alexey S. Sokolov, Fernando Rodriguez, Alexander M. Mazur, Alexey A. Polilov, Richard Benton, Michael B. Evgen’ev, Irina R. Arkhipova, Egor B. Prokhortchouk, Konstantin G. Skryabin
A robotic platform for fluidically-linked human body-on-chips experimentation
Richard Novak, Miles Ingram, Susan Clauson, Debarun Das, Aaron Delahanty, Anna Herland, Ben M. Maoz, Sauveur S. F. Jeanty, Mahadevabharath R. Somayaji, Morgan Burt, Elizabeth Calamari, Angeliki Chalkiadaki, Alexander Cho, Youngjae Choe, David Benson Chou, Michael Cronce, Stephanie Dauth, Toni Divic, Jose Fernandez-Alcon, Thomas Ferrante, John Ferrier, Edward A. FitzGerald, Rachel Fleming, Sasan Jalili-Firoozinezhad, Thomas Grevesse, Josue A. Goss, Tiama Hamkins-Indik, Olivier Henry, Chris Hinojosa, Tessa Huffstater, Kyung-Jin Jang, Ville Kujala, Lian Leng, Robert Mannix, Yuka Milton, Janna Nawroth, Bret A. Nestor, Carlos F. Ng, Blakely O’Connor, Tae-Eun Park, Henry Sanchez, Josiah Sliz, Alexandra Sontheimer-Phelps, Ben Swenor, Guy Thompson II, George J. Touloumes, Zachary Tranchemontagne, Norman Wen, Moran Yadid, Anthony Bahinski, Geraldine A. Hamilton, Daniel Levner, Oren Levy, Andrzej Przekwas, Rachelle Prantil-Baun, Kevin K. Parker, Donald E. Ingber
High-density spatial transcriptomics arrays for in situ tissue profiling
Sanja Vickovic, Goekcen Eraslan, Fredrik Salmen, Johanna Klughammer, Linnea Stenbeck, Tarmo Aijo, Richard Bonneau, Jose Fernandez Navarro, Ludvig Bergenstraahle, Joshua Gould, Mostafa Ronaghi, Jonas Frisen, Joakim Lundeberg, Aviv Regev, Patrik L Staahl
The mesoSPIM initiative: open-source light-sheet mesoscopes for imaging in cleared tissue
Fabian F. Voigt, Daniel Kirschenbaum, Evgenia Platonova, Stéphane Pagès, Robert A. A. Campbell, Rahel Kästli, Martina Schaettin, Ladan Egolf, Alexander van der Bourg, Philipp Bethge, Karen Haenraets, Noémie Frézel, Thomas Topilko, Paola Perin, Daniel Hillier, Sven Hildebrand, Anna Schueth, Alard Roebroeck, Botond Roska, Esther Stoeckli, Roberto Pizzala, Nicolas Renier, Hanns Ulrich Zeilhofer, Theofanis Karayannis, Urs Ziegler, Laura Batti, Anthony Holtmaat, Christian Lüscher, Adriano Aguzzi, Fritjof Helmchen
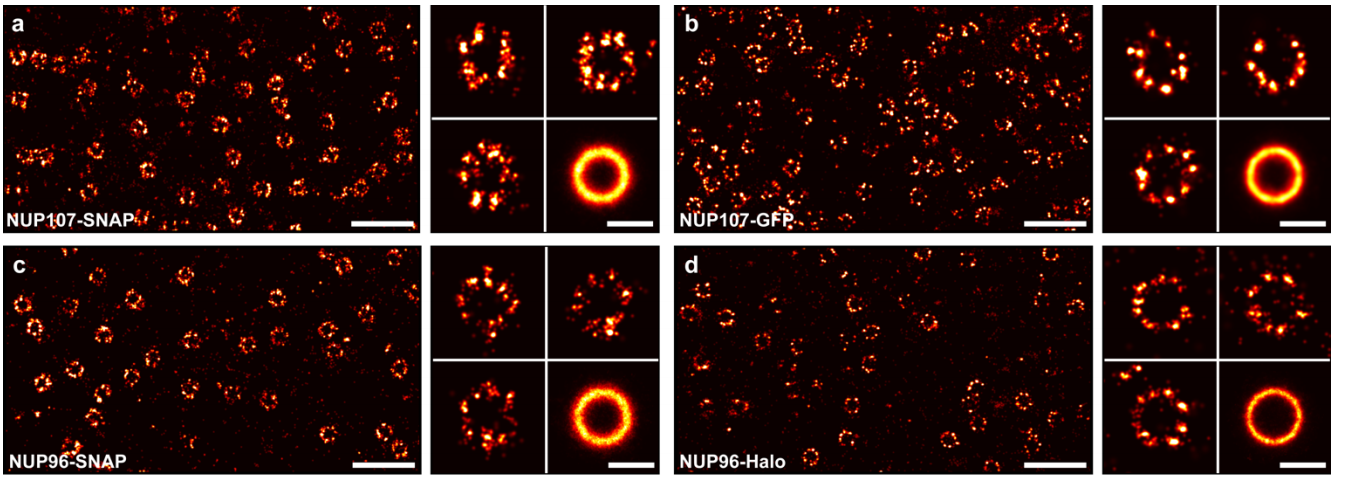
Nuclear pores as versatile reference standards for quantitative superresolution microscopy
Jervis Vermal Thevathasan, Maurice Kahnwald, Konstanty Cieśliński, Philipp Hoess, Sudheer Kumar Peneti, Manuel Reitberger, Daniel Heid, Krishna Chaitanya Kasuba, Sarah Janice Hoerner, Yiming Li, Yu-Le Wu, Markus Mund, Ulf Matti, Pedro Matos Pereira, Ricardo Henriques, Bianca Nijmeijer, Moritz Kueblbeck, Vilma Jimenez Sabinina, Jan Ellenberg, Jonas Ries
The Fruit Fly Brain Observatory: From Structure to Function
Nikul H. Ukani, Chung-Heng Yeh, Adam Tomkins, Yiyin Zhou, Dorian Florescu, Carlos Luna Ortiz, Yu-Chi Huang, Cheng-Te Wang, Mehmet K. Turkcan, Tingkai Liu, Paul Richmond, Chung-Chuan Lo, Daniel Coca, Ann-Shyn Chiang, Aurel A. Lazar
Comparing quality of reporting between preprints and peer-reviewed articles in the biomedical literature
Clarissa F. D. Carneiro, Victor G. S. Queiroz, Thiago C. Moulin, Carlos A. M. Carvalho, Clarissa B. Haas, Danielle Rayêe, David E. Henshall, Evandro A. De-Souza, Felippe Espinelli, Flávia Z. Boos, Gerson D. Guercio, Igor R. Costa, Karina L. Hajdu, Martin Modrák, Pedro B. Tan, Steven J. Burgess, Sylvia F. S. Guerra, Vanessa T. Bortoluzzi, Olavo B. Amaral
Ten myths around open scholarly publishing
Jonathan P Tennant, Harry Crane, Tom Crick, Jacinto Davila, Asura Enkhbayar, Johanna Havemann, Bianca Kramer, Ryan Martin, Paola Masuzzo, Andy Nobes, Curt Rice, Bárbara S Rivera-López, Tony Ross-Hellauer, Susanne Sattler, Paul Thacker, Marc Vanholsbeeck
Applicants are sought for two 3-year doctoral contracts to work on developmental variability and canalization in Ascidians.
Context:
Within each animal species, embryonic development is highly reproducible, ensuring the production of a complex organism with precisely arranged and shaped organs and tissues. This constancy of embryogenesis against genetic polymorphism and fluctuating environmental conditions is critical for the perpetuation of the species, and has been referred to as developmental canalization (Waddington, 1942).
Among animals, nematodes and ascidians (Lemaire, 2011), exemplify the most extreme form of developmental canalization: the quasi-invariant behaviour of individual embryonic cells, such that each cell can be named unambiguously and found across individuals of the same or related species. Such organisms constitute attractive models to unravel the still elusive molecular basis of canalization.
The two proposed projects will explore how these variations are buffered, or canalized, in one class of animals with a highly stereotyped embryogenesis, the ascidians (Lemaire, 2011). The links will bring you to the project page of the CBS2 doctoral school.
Candidates are encouraged to contact by May 1st the future supervisors: patrick.lemaire@crbm.cnrs.fr (both projects) and gregoire.malandain@inria.fr (computer science project) by sending a CV with exam rankings, a motivation letter and the e-mail of two potential academic referees. Application deadline of selected candidates to the doctoral schools will be May 9
We will focus on the FGF/Ras/ERK pathway, the major fate-inducing pathway during early ascidian embryogenesis. Using a fluorescent reporter (Cova et al., 2017) and lightsheet imaging, we will first quantify in live embryos the variability of the dynamics of the activation downstream component of the pathway, ERK, between individuals of the same species and between species, during normal development. This initial set of measurements will be used as a reference to identify molecular or environmental perturbations that increase or decrease the variability of ERK signalling.
We will first test Waddington’s “canalization” proposal that any perturbation of wild-type development increases phenotypic variance (Waddington, 1942). We will then analyse the effect of changes in temperature and salinity on ERK signalling precision, and test the hypothesis that the Hsp90 chaperone acts as a phenotypic buffering mechanism during development (Zabinsky et al., 2018).
Finally, we will test the hypothesis that the precision of ERK activation may result from positive or negative feedback loops acting within the FGF/ras/ERK signalling pathway. Taken together the results obtained during this project will establish the ascidian embryos as a paradigm to study phenotypic robustness during development.
(co-supervised with Grégoire Malandain, INRIA, Sophia Antipolis)
The aim of this computational biology project is to develop a conceptual framework to quantify developmental variability, with single cell resolution, within and between ascidian species and to use this framework to explore the spatio-temporal structure of this variability.
The starting point of this project is the availability of a collection of 12 digitalized wild-type embryos in which each cell was systematically segmented and tracked every two minutes for up to 5 cell divisions during embryogenesis. This digitalization allowed to associate to each individual cell a set of properties defining their embryological (fate), geometrical (volume, surface) and topological (neighborhoods and surface of contact with neighbors) features (Guignard et al., 2018). For an example see our work among the Node’s Xmas GIFs 2018.
To characterize the temporal and spatial structure of the developmental variability of these embryos, we propose to construct a conceptual framework describing average ascidian development and to quantify deviations from this average.
For this, we will construct a toolbox that will make it possible to: 1) Align digital embryos in time and space; 2) Construct average digital embryos for a given (sub)population and/or species; 3) Characterize the geometric and topological variability of a population of 3D shapes.
We will use this toolbox to address two different types of issues. The first is of biological nature and relates to the expected pattern of variability across time. The second is more methodological, and aims at improving our current segmentation strategy by including knowledge gained from previously-segmented embryos.
References:
Cova, C. de la, et al. (2017). A Real-Time Biosensor for ERK Activity Reveals Signaling Dynamics during C. elegans Cell Fate Specification. Dev. Cell 42, 542-553.e4.
Guignard, L., Fiuza, U.-M., et al. (2018). Contact-dependent cell communications drive morphological invariance during ascidian embryogenesis. bioRxiv 238741.
Lemaire, P. (2011). Evolutionary crossroads in developmental biology: the tunicates. Development 138, 2143–2152.
Waddington, C. H. (1942). canalization of development and the inheritance of acquired characters. Nature 150, 563–565. Zabinsky, R. A., et al. (2018). It’s not magic – Hsp90 and its effects on genetic and epigenetic variation. Semin. Cell Dev. Biol.
In this episode from our series exploring 100 ideas in genetics, we’re telling the often-overlooked stories of four women who helped to shape the science of life. We pick four women whose contributions to the history of 20th century genetics have been obscured by sexist scientific culture: Esther Lederberg, Harriet Creighton, Tsuneko Okazaki and Martha Chase.
If you enjoy the show, please do rate and review and spread the word. And you can always send feedback and suggestions for future episodes and guests to podcast@geneticsunzipped.com
Investigating basic principles of tumor hierarchy in Drosophila
Tumors are often heterogeneous, composed by different cell types that exhibit a hierarchical organization reminiscent to developing tissues. At the top of the hierarchy lie Cancer Stem Cells (CSCs) that have the ability to undergo unlimited self-renewal, to propagate tumor growth, but also to differentiate in different cell types to generate cellular heterogeneity. The balance between self-renewal and differentiation determines the speed of tumor growth as well as the proportion of CSCs. The mechanisms that instruct CSCs to self-renew or differentiate are unclear, but it is suspected that the redeployment of developmental programs could be involved.
In the lab, we take advantage of our deep understanding of how the Drosophila brain is built during development to explore how perturbation of these mechanisms can trigger tumorigenic growth. We combine single-cell transcriptomics, numerical modeling and the powerful genetic tools available in Drosophila to investigate how coopted neuro-developmental programs regulate the growth and heterogeneity of neural tumors. Using these approaches, we have uncovered several signaling pathways that are specifically active in a sup-population of CSCs in a model of neural tumors. Such pathways are conserved in mammals but their role in cancer is unclear.
The aim of the project is to decipher how genetic manipulations of these pathways affect the population of CSCs and tumor growth. This will involve generating new Drosophila transgenic lines, immunochemistry, confocal microscopy as well as new single cell transcriptomic approaches. We hope to provide new perspectives for the elimination or control of CSC populations in human.
Scientific background. A long neglected component of the tumor microenvironment are the neurons of the peripheral nervous system. The presence of neural fibers in tumors is now well established but their origin and functional impact on disease progression remain poorly understood. Our team is studying how the different branches of the peripheral nervous system (sympathetic, parasympathetic, enteric and sensory) contribute to the development and spread of pancreatic cancer (PC). Our recent work has revealed significant plasticity and remodeling of the sympathetic fibers that innervate pre-tumor and tumor lesions in mouse models of PC.
Objectives. The main objective of this PhD will be to study the molecular signaling pathways that regulate tumor-induced neuronal plasticity and to test how interfering with this process may affects tumor growth and metastatic progression.
Methods. The team developed new methods for quantitative analysis of neural networks in the entire mouse pancreas. These approaches combine whole-organ clearing and 3D imaging by light sheet fluorescent microscopy (LSFM). An image analysis workflow has been set-up to calculate parameters describing neural network structures and interactions with the main cellular components of the tumor microenvironment. Candidate signaling pathways, including neurite outgrowth and axon guidance pathways, will be identified and validated in vivo using neutralizing antibodies. Tumor growth and the occurrence of metastasis will be investigated by histopathological analysis and bioluminescence imaging.
Expected results. The results of the study will shed new light on the interactions between the nervous system and tumors and will identify potential therapeutic targets for modulating tumor innervation in PC.
Candidate profile. The project is at the intersection of neurobiology and oncology. We are looking for a highly motivated candidate with a solid scientific understanding of molecular and cellular biology and a good knowledge of statistics for biological research.
Crosstalk between cancer and immune cells in triple-negative breast cancer: uncover molecular mechanisms to design molecular therapies.
Topics. This PhD project has been designed as a co-direction thesis between CRCM/IPC (Borg-Team) and IBDM (Maina-Team) to study molecular and biological aspects linked to Triple-Negative Breast Cancer (TNBC). It is based on the use of a unique mouse genetic setting (MMTV-R26Met) recapitulating several molecular/biological TNBC features, including: a) intra-/inter-tumoural heterogeneity; b) resistance to treatments used in clinic; c) lung metastasis. Moreover, MMTV-R26Met TNBC display inflammatory background with infiltrating immune cells, including Tumour-Infiltrating-Lymphocytes, regulatory-T-cells, cytotoxic-T lymphocytes. These findings together with proteomic/RNA-seq outcomes indicate the existence of a crosstalk between cancer and immune cells recapitulated in MMTV-R26Met TNBC model.
Objectives. The PhD student will apply multi-disciplinary strategies to explore the crosstalk between cancer and immune cells during the formation/evolution of TNBC and its reprogramming during tumour regression following anticancer treatment, focussing on two complementary objectives.
1) An unbiased analysis of screen outcomes to extract molecular components of the cancer-immune cell crosstalk. The student will analyse transcriptome/proteomic data from MMTV-R26Met TNBC model at distinct tumorigenic states to: a) uncover signals belonging to the cancer-immune cell crosstalk; b) follow the dynamics of their expression changes; c) integrate findings with human cancer databases, exploring the existence of patient-subtype signatures. The student will analyse functional relevance of crosstalk components through gain-/loss-of-function studies using MMTV-R26Met TNBC cells.
2) Explore a candidate mechanism implicating the PTK7-EphA2 receptor axis on cancer-immune cell crosstalk, a new putative circuit emerging from Borg Team studies. Interestingly, the MMTV-R26Met TNBC model recapitulates this signalling crosstalk as PTK7 and EphA2 are upregulated in tumours. The student will analyse this circuit in cancer and immune cells both in MMTV-R26Met TNBC and sample patients from IPC. Moreover, studies will highlight modulation of this circuit during the tumorigenic program and following treatment. Combined therapies targeting both receptors will be tested in vitro/in vivo.
Methodologies. The PhD student will acquire several competences by applying complementary strategies: 1) bioinformatics to analyse screen outcomes and to integrate human cancer databases; 2) histological/FACS analyses to characterize immune cells in tumours; 3) molecular biology, biochemistry, proteomics, to explore cancer-immune cell crosstalk components; 4) gain-/loss-of function studies to assess functionality.
Expected results. Identify the cellular/molecular components of cancer-immune cell crosstalk during tumour onset, evolution, and regression following treatment. Design/test combinatorial treatments in preclinical models combining anticancer drugs with immune-therapy.
Candidate profile. Highly motivated candidate for a challenging project, with strong interest on cancer biology, and interested to apply approaches spanning from cultured cells, tissues analyses, and cancer mouse models. The student will be exposed to a scientific environment that stands out for interdisciplinary expertise.
Molecular control of intercellular junction remodeling during radial intercalation
Scientific background
The insertion of individual mesenchymal cells into an epithelial sheet (radial intercalation) occurs during embryonic development, adult tissue homeostasis, and dissemination of metastatic carcinoma cells. Nevertheless, its control mechanisms remain poorly understood. In particular, little attention has been devoted to the role played by the ‘accepting’ epithelial cells.
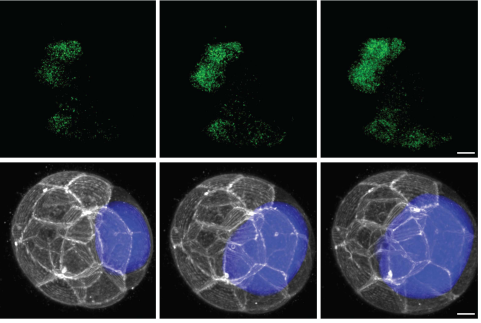
The epithelium of the amphibian Xenopus laevis is a simple and accessible model system to study radial intercalation. During the development of this tissue, mesenchymal multiciliated cells (MCCs) undergo a short apical migration to intercalate into an epithelial layer in correspondence of junctions among at least three cells (aka vertices).
Objectives
In this PhD project, it is proposed to investigate the role played by outer layer epithelial cell cytoskeleton and intercellular junctions in the process of MCC intercalation. We have recently identified the signal triggered by the tyrosine kinase receptor KIT and its ligand SCF as a guidance system that controls the directionality of MCC movements during the intercalation process and their correct homing to outer layer vertices. KIT is expressed in intercalating MCCs, while outer layer cells express a transmembrane form of SCF. The PhD candidate will investigate whether a cell-autonomous signal initiated by SCF plays a role in opening outer layer intercellular junctions to accommodate intercalating MCCs.
Methods
Antibodies and custom–made fluorescent reporter constructs under the control of the outer layer-specific promoter of Xenopus nectin will be used to monitor the status of intercellular junctions and cytoskeletal components at different time points during the process of MCC intercalation through time-lapse confocal microscopy. Constructs coding for dominant-negative forms of cytoskeletal or intercellular junction proteins will be used to assess the role of these structures in MCC intercalation. To explore the possibility that SCF is cell-autonomously required for opening the outer-layer intercellular junctions, we will use nectin promoter-based fluorescent reporter constructs to study the dynamics of cytoskeleton and intercellular junctions following morpholino-mediated depletion of SCF.
Impact
The results obtained will clarify the contribution of epithelial cells to the process of
radial intercalation of mesenchymal cells, and shed new light on the mechanisms underlying signaling by the SCF/KIT pathway during development and metastasis.
Expected profile of the PhD candidate
The PhD candidate must have a strong background in one or several of the following disciplines: cell biology, developmental biology, biochemistry, biophysics. He or she must have a keen interest for quantitative biology and real-time imaging.
Quantitative analysis and simulation of fluid flows powered by cilia in vivo
Scientific background
Ciliated epithelia are present throughout evolution and serve functions ranging from locomotion of marine larvae to mucociliary clearance of pathogens from human airways. These functions are supported by the coordinated beating of myriads of motile cilia at the surface of multiciliated cells (MCCs), which generates robust and regular waves of fluid. The production of such waves depends on multiple parameters integrated across micrometric to millimetric scales, such as MCC density and cilia orientation. These parameters can be adequately studied in the larval mucociliary skin of the amphibian Xenopus, which is organized like the human airway epithelium, but is much easier to manipulate and film. This project will bridge the gap between biology, physics and numerical simulation to bring a new perspective to the understanding of respiratory diseases characterized by altered cilia-driven mucociliary transport, such as COPD or cystic fibrosis.
PhD Objectives
The aim of this interdisciplinary PhD project is to link quantitative experimental analysis to numerical simulation to build a global model of ciliated epithelium organization and activity. The ambition is to generate a multi-parametric model able to explain the emergence of cilia-driven fluid flows at the level of an entire organ. To this aim quantitative live imaging of cilia will be implemented on the 3D Xenopus embryo, which has never been done before. Numerical models will be challenged in silico and through experiments to test their predictive capacity.
Proposed approach
Live imaging: The PhD student will adapt light-sheet microscopy to film fluorescent beating cilia in all MCCs of the Xenopus embryo at various developmental stages.
Image processing: Biophysical analysis will be applied to extract quantitative information regarding cilia beating frequency and orientation, so as to build a global map of the embryo.
Numerical simulation: The quantitative experimental parameters thus acquired will be used to feed Lattice Boltzmann-based simulations, with the aim of generating numerical paradigms to explain the emergence of metachronal ciliary beat waves and spatial flow patterns.
Validation: In silico challenges (changes in ciliated cell density or orientation) will be introduced to predict outcomes on flow patterns, which will be verified through our experimental pipeline.
PhD student’s expected profile
The selected PhD student must have a keen interest in interdisciplinary and quantitative approaches to study biological problems. The ideal candidate will have a solid theoretical background in biological physics. Experience in microscopy techniques and image processing and a strong interest in modeling will be favorably considered.
Role of TSHZ3 gene in the development and function of neuronal systems involved in autism spectrum disorder
State of the art
A collaborative study joining clinical and basic researchers, directed by two teams at the IBDM in Marseille (L. Fasano and L. Kerkerian-Le Goff), has linked an autism spectrum disorder (ASD) syndrome to heterozygous deletions of TSHZ3 gene. This gene codes for the TSHZ3 transcription factor, which is highly expressed in the cerebral cortex projection neurons (CPNs). Heterozygous deletion of Tshz3 in mouse (Tshz3+/-) leads to functional defects at corticostriatal synapses, changes in gene expression pattern as well as behavioral deficits mimicking ASD (reduced social interactions and fields of interest, stereotypies) (Caubit, Gubellini et al Nature Genetics 2016). We have also shown that postnatal Tshz3 deletion produces similar behavioral changes, but different gene expression changes and electrophysiological/synaptic defects at the level of the layer 5 CPNs and the corticostriatal pathway (Chabbert et al, Biological Psychiatry, in press). Interestingly, in this brain circuit TSHZ3 is expressed not only by CPNs but also by striatal cholinergic interneurons (CINs). This raises the issue about the respective role of these two neuronal populations in ASD, and of Tshz3 during pre- and postnatal development. To address this question we have generated several mouse models with conditional Tshz3 deletion.
Objectives
This project aims at elucidating the respective role of Tshz3 loss in CPNs vs. CINs in ASD deficits, and at studying their cellular and molecular substrates.
The specific objectives are:
1) Studying the functional consequences of Tshz3 conditional deletion on the electrophysiological and synaptic properties of CPNs and CINs (excitability, synaptic transmission/plasticity, etc.).
2) Establishing the gene expression profiles of these mice in order to provide a molecular basis for the electrophysiological and behavioral changes.
3) Evaluation whether or not the selective restauration of Tshz3 expression in one of these neuronal populations, in Tshz3+/- mice, can rescue the behavioral, cellular and molecular changes (“rescue” model)
Methods
This project is based on slice electrophysiology (patch-clamp) technique, as well as some immunohistochemistry and molecular biology.
Expected results
We expect that Tshz3 deletion or rescue in CPNs vs. CINs will produce specific changes at behavioral, molecular and electrophysiological level.
Expected candidate profile
The candidate should have a background in Neuroscience and be proficient with slice electrophysiology. Some experience with molecular biology and/or immunohistochemistry is also welcome.
Uncover the interplay between signalling dynamics and cell mechanics during early fate determination of human induced pluripotent stem cells
Human induced pluripotent stem cells (hiPSCs) hold great promise for developmental biology studies and for regenerative medicine owing their ability to generate all cell types of three germ layers (endoderm, mesoderm, and ectoderm). Regulation of hiPSC differentiation relies on dynamic interaction between signalling and physical cues. At the signalling level, spatiotemporally regulated secreted proteins such as BMP, WNT, and ACTIVIN/NODAL induce different fates based on concentration and dynamics of presentation. Yet, physical cues such as cell-cell adhesion, cell shape and geometry, extracellular-matrix mechanics modulate cell fate specification and patterning by altering and/or shaping cellular responses to biochemical signals.
We propose to explore the spatial/temporal dynamic of instructive signals and physical cues governing mesoderm and endoderm lineage acquisition versus neuroectoderm by using hiPSCs down-regulated for the morphogen regulator, GLYPICAN4. The rational arises from our results showing that GLYPICAN4 controls self-renewal versus lineage entry of stem cells by fine-tuning self-renewal signals. We have also evidences for GLYPICAN4 as crucial regulator of neuroectoderm versus mesoderm and endoderm lineage diversification, as illustrated by enhanced propensity of GLYPICAN4 mutant hiPSCs (versus controls) to acquire mesoderm and endoderm fate in response to extracellular signals. Interestingly, undifferentiated GLYPICAN4 mutant hiPSCs display changes in cell-cell and cell-matrix interactions, which are reminiscence of an impaired epithelial-like structure of GLYPICAN4 mutant hiPSCs. Thus, GLYPICAN4 mutant hiPSCs are suitable as model system to explore mechanistically the crosstalk between mechanical properties and signalling processes that drives efficient differentiation of hiPSCs into the three embryonic layers.
For these studies, we will employ the micropatterning technology in which hiPSC, spatially confined onto circle geometry, differentiate to generate simultaneously neuroectoderm, mesoderm, and endoderm organized in adjacent domains with radial pattern. Segregation in these lineages requires quantitative intercalation of BMP, ACTIVIN, WNT signals in a precise temporal dynamics. Thus, we will apply these factors to hiPSC at different concentrations and time. We will analyse differentiation patterns by fluorescence marker imaging with state-of-art microscopy and computational image analysis as well as signal diffusion and receptor patterns at the plasma membrane. Micropatterns will be also used to study physical properties of undifferentiated and differentiating GLYPICAN4 mutant hiPSCs versus controls by performing mechanical measurements (by atomic force microscopy) and by molecular analysis of mechanosignals.
Results from this analysis should highlight how mechanical signals integrate with soluble signals to regulate embryonic specification by differentially modulating cell-cell and cell-matrix interactions and by establishing patterns of differential signal sensitivity in cells.
The candidate is expected to have strong interest in developmental and cell biology as well as expertise in hiPSC technologies.
Investigating the effects of re-expression of the Tshz3 gene in adult mouse brain on autistic behaviours
State of the art
We have established that the heterozygous deletion of the TSHZ3 gene in humans and mice causes autism spectrum disorders (ASD) (Caubit et al, Nat. Genet. 2016). Recently, we have shown that the conditional deletion of Tshz3 in the post-natal cerebral cortex induces autistic behaviours with changes in gene expression and functional defects in cortical projection neurons (CPNs) in layer V (L5) (collaboration with Lydia Kerkerian-Le Goff’s team; Chabbert et al, Biological Psychiatry, under revision). These results point to a critical postnatal function of Tshz3 in maturation and central nervous system (CNS) function, particularly in L5 CPNs. These data encouraged us to generate a Tshz3+/STOP mouse to restore Tshz3 function in a controlled way after crossing with a Cre lineage and test the ability to correct functional defects by restoring Tshz3 expression in the CNS after birth.
Objectives
1) Characterize the behaviour of Tshz3+/STOP heterozygous mice that should exhibit autistic-type behaviours such as Tshz3+/lacZ and Tshz3+/Deltaflox mice already tested.
2) Analyze the behavior of rescue mice produced by crossing Tshz3+/STOP mice with Cre mice (i.e. CamK2-Cre).
3) Identify the molecular pathways modified by the postnatal loss of Tshz3 and restored after “rescue” (RNAseq transcriptomic analysis).
4) Analyze the behavioral, molecular and functional consequences of the loss of Tshz3 function specifically in the CPNs of L5.
5) Identify the direct targets of the transcription factor TSHZ3 by in vivo chromatin immunoprecipitation approaches (ChIP; ChIP-seq).
Methods
This project focuses on genetic, transcriptomic and immunohistochemistry approaches (in vivo target validation). The functional analysis by electrophysiology will be carried out in collaboration with Lydia Kerkerian-Le Goff’s team.
Expected results
This project will determine 1) whether restoring Tshz3 expression after birth improves the behavior of Tshz3+/STOP mice; 2) identifying for the first time direct targets of TSHZ3; 3) identifying molecular targets relevant for the development of future therapeutic strategies for ASDs; and 4) specifying the contribution of Tshz3 loss of function in L5 CPNs to characteristic ASD phenotypes.
Expected candidate profile
The candidate should have training in neurobiology and practical experience in one of the techniques planned for the thesis. An experiment in mouse genetics would be beneficial but not mandatory.
impact of gut microbiota on host behavior and physiology
Scientific background
It is now very well established that gut-associated bacteria can impact the behavior and the physiology of their eukaryotic host. The PhD thesis project is aimed at using the powerful genetic tools available in the Drosophila model and the relative simplicity of its gut microbiota to study, at the molecular level, the molecular dialog between the microbiota and its host. In two recent publications, (Kurz et al, Elife, 2017: Charroux et al, Cell Host Microbe, 2018), the lab has shown that a metabolite produced by gut-associated bacteria, called peptidoglycan, can cross the gut epithelium and reach the insect blood where it interferes with various organs (fat body, ovaries, brain…) and modifies functions (behavior changes, organ wasting…). The PhD student will use the newest genome editing technologies (Crispr…), genetic tools and latest imaging microscopy technics to dissect the precise cellular and molecular mechanisms of the dialog that exist between gut-resident bacteria and some specific cells of the host. Recent results showing that mice deficient in peptidoglycan-sensing proteins exhibit social behavioral alterations suggest that the mechanisms that we study in Drosophila also exist in mammals.
Profile of the candidate
We look for an enthusiastic and ambitious student with a strong interest in the genetics of host-bacteria interactions. The candidate is expected to have a background in molecular biology and should hold a Master Degree in Bioscience Engineering, Biotechnology or Biology. The candidate should have a level of proficiency in English which is sufficient to communicate effectively with colleagues.
We seek an outstanding postdoctoral candidate to join the Yeh Laboratory at Texas A&M University in College Station (http://biomed.tamu.edu/tml). Our group uses interdisciplinary and quantitative approaches to study the molecular and cellular basis of embryonic development with specific focus on brain development. In this context, we are interested in understanding how complex multi-enhancer regulatory landscapes interact with gene promoters through the application of super-resolution, live cell imaging. A particular strength of the lab is the development of custom, state-of-the-art microscopy systems for applications in the life sciences.
The candidate will be self-motivated, creative, and able to work collaboratively with a group of researchers with expertise in microscopy, physics, and developmental biology. A PhD in the biological sciences (or related fields) with at least 3 years of laboratory research experience in zebrafish developmental biology is required. Experience with quantitative imaging, in addition to experience in zebrafish development, will be considered positively, but is not required.
This is a NIH-funded position with full benefits. This position is renewable based upon good performance of the candidate and availability of funds. Salary will be competitive and dependent on the level of experience of the candidate. This position is currently open, and applications will be accepted until position is filled.
Applicants should provide,
cover letter describing research interests and career goals,
CV,
name and contact information from at least two references.
This is the latest dispatch from a recipient of a Development Travelling Fellowship, funded by our publisher The Company of Biologists.
Learn more about the scheme, including how to apply, here, and read more stories from the Fellows here.
Estefanía Sánchez-Vásquez
(Peruvian woman doing a PhD in Argentina)
Lately there is much discussion of what an exciting time it is to be a Developmental Biologist. Technologies such as CRISPR allow us to precisely modify diverse and “non-genetic” organisms, enabling understanding of the function of several genes in complex organism. Single cell analysis helps us understand heterogeneity, differentiation and identification of several new cell types. However, in some parts of Latin America such as Brazil and Argentina, state politics are cutting funding to science. In Peru, as in other Latin American countries, studying Biology is viewed as impractical and raises concern to our parents: Will you get a job? Will you make money? Are you going to be a school teacher? Even given these harsh realities, some people passionately choose to do science. To quote Frances Arnold, winner of the Nobel Prize in Chemistry, 2018: “It just comes! I love what I do and I enjoyed every day”. In this context, I write this note to say thank you for the Development travelling fellowship from the Company of Biologists which, without any discrimination by nationality or gender, provides a unique opportunity to travel to other labs to do new experiments and learn techniques.
In the middle of work at the egg bench.
This fellowship allowed me to travel from Chascomús (Argentina) to Pasadena (USA) to Marianne Bronner’s lab at Caltech, to finish the project that we started in Argentina about a tumor-suppressor microRNA, miR-203. In brief, miR-203 is epigenetically downregulated to allow the migration of the neural crest cells. Working with microRNAs, neural crest cells and chicken embryos in two different contexts (INTECh* and CALTECH) makes me realize how lucky and grateful I am with the professors that decided to do science, even in “impractical” situations, because without them I would not be writing this note. Happily, science does not discriminate and information is available for everyone. This opportunity was life-changing, not only because I had the chance to be in one of the best chicken labs but also because I met great people that will always be my friends. I also want to thank the Bronner Lab for confirming to me that science is more than papers; it is about people that are doing what they love and are inspired by the questions they want to answer, and that does not change even if you are in different parts of the world.
Estefanía has a paper out in press in Development – coauthored with Marianne Bronner, whose lab she visited as part of the Travelling Fellowship. You can check it out here:
A postdoctoral position is currently available in the Pujol_Ewbank Laboratory, part of the Labex INFORM and Turing Centre for Living Systems (CENTURI), to study the cellular and molecular response to damage, or how wound healing and immune responses are coordinated in a single cell.
Description: We are looking for a highly motivated, enthusiastic and interactive postdoctoral candidate to join our multidisciplinary research team. The ideal candidate should have a strong background in Cell biology and Genetics. The research in our lab is focused on studying the molecular and cellular mechanisms underlying the response to damage in the C. elegans epidermis. This involves deciphering the chemical and mechanical triggers and understanding the links between signalling and cytoskeletal remodelling. In our work, we use in vivo imaging to capture cell biological changes provoked by injury, combined with genetic, genomic and biophysical approaches. Experience in invertebrate model genetics is desirable but not necessary. The successful postdoc candidate will receive training in genetics, molecular and cell biology approaches for C. elegans research.
The position is available for 2 years with the possibility to apply for extension. Interested candidates should send a CV, a description of research experience and contact information for two references to Nathalie Pujol pujol[at]ciml.univ-mrs.fr
Selected publications:
Taffoni C. et al. 2019. Microtubule plus-end dynamics link wound repair to the innate immune response. BioRXiv 512632.
Dodd W. et al. 2018. A Damage Sensor Associated with the Cuticle Coordinates Three Core Environmental Stress Responses in C. elegans. Genetics 208, 1467-1482.
Zugasti O. et al. 2016. A quantitative genome-wide RNAi screen in C. elegans for antifungal innate immunity genes. BMC Biol 14, 35.
Kim K.W. et al. 2016. Coordinated inhibition of C/EBP by Tribbles in multiple tissues is essential for C. elegans development. BMC Biol 14, 104.
Ewbank J.J. and Pujol N. 2016. Local and long-range activation of innate immunity by infection and damage in C. elegans. Curr Opin Immunol 38, 1-7.
Zugasti O. et al 2014. Activation of a G protein-coupled receptor by its endogenous ligand triggers the innate immune response of C. elegans. Nat Immunol 15, 833-838.
Rouger V. et al. 2014. Independent Synchronized Control and Visualization of Interactions between Living Cells and Organisms. Biophysical journal 106, 2096-2104.
Dierking K. et al. 2011. Unusual regulation of a STAT protein by an SLC6 family transporter in C. elegans epidermal innate immunity. Cell Host Microbe 9, 425-435.
 (3 votes)
(3 votes)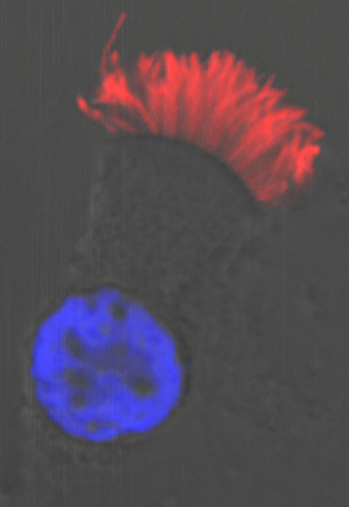









 (No Ratings Yet)
(No Ratings Yet)

{kind=link}