BSDB Gurdon Summer Studentship Report (16)
Posted by BSDB, on 2 November 2017
![]() Established by the British Society for Developmental Biology in 2014, The Gurdon/The Company of Biologists Summer Studentship scheme provides financial support to allow highly motivated undergraduate students an opportunity to engage in practical research during their summer vacation. Each year, ten successful applicants spend eight weeks in the research laboratories of their choices, and the feedback we receive is outstanding.
Established by the British Society for Developmental Biology in 2014, The Gurdon/The Company of Biologists Summer Studentship scheme provides financial support to allow highly motivated undergraduate students an opportunity to engage in practical research during their summer vacation. Each year, ten successful applicants spend eight weeks in the research laboratories of their choices, and the feedback we receive is outstanding.
Our fifth report from the 2017 group of student awardees comes from Jake Cornwall Scoones (student at University of Cambridge), who undertook his studentship with Anna Philpott at the Dept. of Oncology in Cambridge.
Transdifferentiation of pancreatic organoids
 This summer, I had the amazing opportunity of undertaking a Gurdon Summer Studentship, working in Prof. Anna Philpott’s lab, under the guidance of Dr. Roberta Azzarelli at the Cambridge Oncology Department. Having completed first-year Natural Sciences, I was intrigued to learn more about the molecular mechanisms that underpin development. Focused on a potential transdifferentiation pathway in pancreatic cells, the project has been both fascinating and informative, teaching me many invaluable skills, from tissue culture, to growing organoids, to having the patience required for many of the complex procedures involved.
This summer, I had the amazing opportunity of undertaking a Gurdon Summer Studentship, working in Prof. Anna Philpott’s lab, under the guidance of Dr. Roberta Azzarelli at the Cambridge Oncology Department. Having completed first-year Natural Sciences, I was intrigued to learn more about the molecular mechanisms that underpin development. Focused on a potential transdifferentiation pathway in pancreatic cells, the project has been both fascinating and informative, teaching me many invaluable skills, from tissue culture, to growing organoids, to having the patience required for many of the complex procedures involved.In recent years, many have championed regenerative medicine as a solution to diseases associated with cell loss like diabetes mellitus. This technique, where cells are grown and differentiated to replace dead or diseased cells, has the potential to overcome many of the pitfalls of transplantation, most notably immune rejection. To minimise rejection, inserted cells must display high genomic identity with the recipient. These cells can be derived by one of two methods:1, 2 through a differentiation programme, transitioning embryonic(-like)3 stem cells into the fate desired; or via transdifferentiation, through introducing factors that tweak the cell’s genome and epigenome.
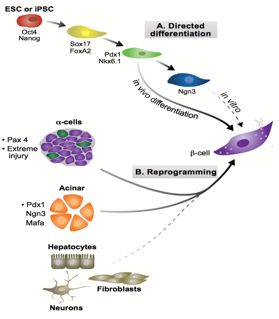
During development, cell-lineages sequentially accrue epigenetic modifications that consign them to their fate, transforming from a totipotent zygote to terminally differentiated cells. Waddington’s visual metaphor, envisaging this sequential differentiation as a ball rolling down a terrain of bifurcating valleys, helps to frame questions in developmental biology.4 Upon transplanting the nucleus of somatic Xenopus cells into enucleated eggs, Gurdon found that the resultant cell was pluripotent, capable of differentiating into all cell types.5 In so doing, Gurdon demonstrated that factors within an egg have the capacity to modify the epigenetic state of a nucleus, implying that the ball can roll back up the valley. In the case of pancreatic transdifferentiation, with sufficient knowledge of this terrain and with the correct molecular intervention, we should able to push the ball into our chosen valley.
Pancreatic development proceeds through two stages: the primary transition (embryonic day 9 (E9) to E12.5); and the secondary transition (E12 to birth).6 The primary transition sees endodermal thickening, followed by the pancreatic progenitor proliferation leading to pancreatic bud formation. Through a process of epithelial stratification and micro-lumen formation, the initial tubular morphology that will later characterise the pancreas begins to emerge. This initial expansion phase is followed by rounds of specification and patterning, forming a bipotent trunk and a multipotent tip. Adult pancreata are comprised of three different cell types, namely acinar, duct and endocrine cells, the former derived from tip progenitors and the latter two from trunk cells.
Endocrine development, part of the secondary transition, is triggered by the transient expression Neurogenin 3 (Ngn3) above a threshold level, and proceeds through delamination from the epithelium and aggregation into Islets of Langerhans. Ngn3+ cells are specified to one of five fates in a temporally regulated manner:7 α-cells produced upon earliest Ngn3-activation synthesising glucagon; followed by β-cells producing insulin; then δ-cells producing somatostatin; then PP-cells producing pancreatic polypeptide; and finally ε-cells producing ghrelin. Previous work from the lab by Azzarelli et al.8 demonstrated Ngn3 is phosphoregulated by proline-directed kinases (e.g. CDKs), regulating the balance between proliferation, (Ngn3 hyper-phosphorylation), and differentiation, (Ngn3 under-phosphorylation). β-cell maturation is promoted by the transcriptional regulator Pdx1, among others, whose expression remains confined to β-cells in the adult pancreas. Working in tandem with Pdx1, MafA transcriptionally regulates genes involved in insulin secretion and biosynthesis and it is thus considered a marker of mature β-cell identity.
Ngn3, crucial in assigning endocrine fate initially, and MafA and Pdx1, important in β-cell maturation, have been previously over-expressed in combination to induce transdifferentiation of acinar cells to β-fate in an in vivo model. These factors were used in our experiment to see if such transdifferentiation is possible in an in vitro model, specifically pancreatic organoids, a form of 3D cell culture grown in matrigel. Upon over-expression of these three factors, it is possible to test for changes in cell fate, by looking at symptomatic traits of β-cells, namely the production of insulin.
Over-expression can be achieved through the infection of organoids with lentiviral vectors carrying genetic loci that upon infection will be inserted into the cells’ nuclear genomes. The activation of these imported genes should be controllable so a transactivator is used which, when bound to the antibiotic doxycycline (Dox), becomes active, binding the promoter of our genes of interest, inducing their transcription. The three factors are necessarily expressed together through their transcription within a single ORF, containing MafA, Pdx, Ngn3 and GFP (a fluorescent marker) sequences, separated by loci encoding the cleavage peptide 2A, meaning upon translation, cleavage ensues resulting in four functional proteins. Hence a GFP signal is sufficient to determine the expression of all three factors, leaving other wavelengths free to test for pancreatic hormones through immunohistochemistry.
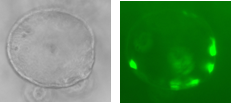
Two lentiviral vectors were used to infect organoids, one carrying the transactivator gene Tet3G, and the other carrying the locus of interest, Pdx-MafA-Ngn3-GFP (PMN), or GFP as our control. Viruses were produced by the transfection of HEK cells with several plasmids, each containing genes encoding different viral proteins plus the locus of interest, followed by incubation, viral purification and titration. Organoids were incubated with viruses for a week with Dox and then fixed with PFA. Immunohistochemistry was performed, staining organoids for insulin. Preliminary results are very encouraging and I look forward to seeing how my contribution fits into the larger project as a whole.
This studentship has been an invaluable experience, allowing me to gain an understanding of real-world lab science, a practice venturing into the unknown, far from the rushed experiments for which answers were already known performed as a part of my course. I have developed lab-skills, most notably having the rare opportunity to work with organoids, a relatively novel technology. I urge other students to consider applying for this fantastic opportunity in future years.
References
- Pagliuca, Felicia W., and Douglas A. Melton. “How to make a functional β-cell.” Development 140.12 (2013): 2472-2483.
- Zhou, Qiao, and Douglas A. Melton. “Extreme makeover: converting one cell into another.” Cell Stem Cell 3.4 (2008): 382-388.
- Takahashi, Kazutoshi, and Yamanaka. “Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors.” Cell 126.4 (2006): 663-676.
- Waddington, C. H. (1957). The Strategy of the Genes. London : George Allen & Unwin
- Gurdon, John B., Thomas R. Elsdale, and Michel Fischberg. “Sexually mature individuals of Xenopus laevis from the transplantation of single somatic nuclei.” Nature 182.4627 (1958): 64-65.
- Benitez, Cecil M., William R. Goodyer, and Seung K. Kim. “Deconstructing pancreas developmental biology.” Cold Spring Harbor Perspectives in Biology 4.6 (2012): a012401.
- Johansson, Kerstin A., et al. “Temporal control of neurogenin3 activity in pancreas progenitors reveals competence windows for the generation of different endocrine cell types.” Developmental Cell 12.3 (2007): 457-465.
- Azzarelli, Roberta, et al. “Multi-site Neurogenin3 Phosphorylation Controls Pancreatic Endocrine Differentiation.” Developmental Cell 41.3 (2017): 274-286.
 (1 votes)
(1 votes)
 (No Ratings Yet)
(No Ratings Yet)
 Named after the mythical “Land of the young”, Tír na nÓg, Nanog is a homeobox transcription factor expressed in embryonic stem cells (ESCs) aiding continual cellular proliferation alongside the other main pluripotency factors, Oct4 and Sox2. However, despite its contribution to pluripotency, much is still unknown of the mechanistic roles that Nanog plays within stem cells, since cells kept in culture deficient in Nanog retain the ability to self-renew (
Named after the mythical “Land of the young”, Tír na nÓg, Nanog is a homeobox transcription factor expressed in embryonic stem cells (ESCs) aiding continual cellular proliferation alongside the other main pluripotency factors, Oct4 and Sox2. However, despite its contribution to pluripotency, much is still unknown of the mechanistic roles that Nanog plays within stem cells, since cells kept in culture deficient in Nanog retain the ability to self-renew (

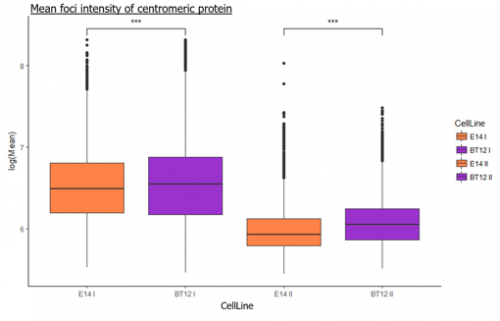

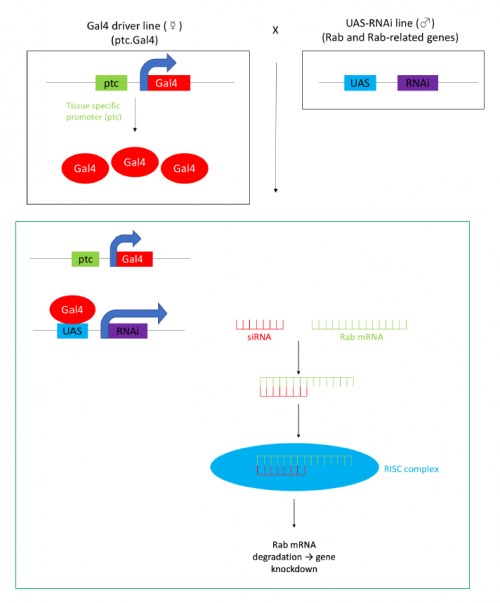
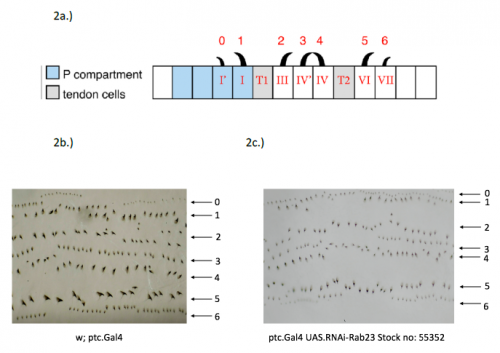
 The tectorial membrane (TM) is an extracellular matrix (ECM) that overlies the organ of Corti in the inner ear and is crucial for our sense of hearing. It is composed of collagen fibrils embedded in a tectorin-based matrix. The precise alignment of the collagen fibrils across the TM is a feature considered critical for hearing, but very little is known about how this pattern is generated. On p.
The tectorial membrane (TM) is an extracellular matrix (ECM) that overlies the organ of Corti in the inner ear and is crucial for our sense of hearing. It is composed of collagen fibrils embedded in a tectorin-based matrix. The precise alignment of the collagen fibrils across the TM is a feature considered critical for hearing, but very little is known about how this pattern is generated. On p.  Embryonic patterning is dependent on the establishment of the anteroposterior (AP) and dorsoventral axes early in development. In mammalian embryos, these axes are established by a breaking of symmetry in the epiblast, which involves signals from the extra-embryonic tissues. However, the molecular mechanisms that control this process are still not fully understood. On p.
Embryonic patterning is dependent on the establishment of the anteroposterior (AP) and dorsoventral axes early in development. In mammalian embryos, these axes are established by a breaking of symmetry in the epiblast, which involves signals from the extra-embryonic tissues. However, the molecular mechanisms that control this process are still not fully understood. On p.  The core autophagy protein Atg16L1 has been identified as a genetic risk factor in inflammatory bowel disease, but how it plays this role has remained unclear. On p.
The core autophagy protein Atg16L1 has been identified as a genetic risk factor in inflammatory bowel disease, but how it plays this role has remained unclear. On p.  Christiane Nüsslein-Volhard is Director Emeritus at the Max Planck Institute for Developmental Biology in Tübingen, Germany. In 1995, she was awarded the Nobel Prize for Physiology and Medicine, along with Eric Wieschaus and Edward Lewis, for her work on the genetic control of embryogenesis using the fruit fly Drosophila melanogaster. In the 1990s, she transitioned her lab to working with zebrafish (Danio rerio), using similar forward genetic approaches to those that had proved so successful in Drosophila to uncover key regulators of vertebrate development. We met with Christiane at the recent International Society for Developmental Biology (ISDB) meeting in Singapore, to talk about her research, the impact of the Nobel Prize and the challenges of being a ‘woman in science’. See the
Christiane Nüsslein-Volhard is Director Emeritus at the Max Planck Institute for Developmental Biology in Tübingen, Germany. In 1995, she was awarded the Nobel Prize for Physiology and Medicine, along with Eric Wieschaus and Edward Lewis, for her work on the genetic control of embryogenesis using the fruit fly Drosophila melanogaster. In the 1990s, she transitioned her lab to working with zebrafish (Danio rerio), using similar forward genetic approaches to those that had proved so successful in Drosophila to uncover key regulators of vertebrate development. We met with Christiane at the recent International Society for Developmental Biology (ISDB) meeting in Singapore, to talk about her research, the impact of the Nobel Prize and the challenges of being a ‘woman in science’. See the  During development, genes are transcribed at specific times, locations and levels. In recent years, the emergence of quantitative tools has significantly advanced our ability to measure transcription with high spatiotemporal resolution in vivo. Here, Angela DePace and co-workers highlight recent studies that have used these tools to characterize transcription during development, and discuss the mechanisms that contribute to the precision and accuracy of the timing, location and level of transcription. See the
During development, genes are transcribed at specific times, locations and levels. In recent years, the emergence of quantitative tools has significantly advanced our ability to measure transcription with high spatiotemporal resolution in vivo. Here, Angela DePace and co-workers highlight recent studies that have used these tools to characterize transcription during development, and discuss the mechanisms that contribute to the precision and accuracy of the timing, location and level of transcription. See the  Cortical interneurons are a diverse group of neurons that project locally and are crucial for regulating information processing and flow throughout the cortex. Recent studies in mice have advanced our understanding of how these neurons are specified, migrate and mature. Here, John Rubenstein and colleagues evaluate new findings that provide insights into the development of cortical interneurons and that shed light on when their fate is determined, on the influence that regional domains have on their development, and on the role that key transcription factors and other crucial regulatory genes play in these events. See the
Cortical interneurons are a diverse group of neurons that project locally and are crucial for regulating information processing and flow throughout the cortex. Recent studies in mice have advanced our understanding of how these neurons are specified, migrate and mature. Here, John Rubenstein and colleagues evaluate new findings that provide insights into the development of cortical interneurons and that shed light on when their fate is determined, on the influence that regional domains have on their development, and on the role that key transcription factors and other crucial regulatory genes play in these events. See the  The 2nd NEUBIAS school for Bioimage Analysts will be organized in Jan. 2018 in Szeged, Hungary, and the registration is now open (Organizers: Jean-Yves Tinevez & Kota Miura). Please visit the linked URL below for more details. This school is the most advanced among three levels of NEUBIAS school. Deadline for the registration is Nov. 9th.
The 2nd NEUBIAS school for Bioimage Analysts will be organized in Jan. 2018 in Szeged, Hungary, and the registration is now open (Organizers: Jean-Yves Tinevez & Kota Miura). Please visit the linked URL below for more details. This school is the most advanced among three levels of NEUBIAS school. Deadline for the registration is Nov. 9th.