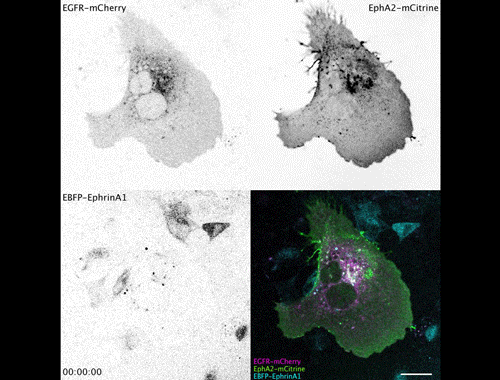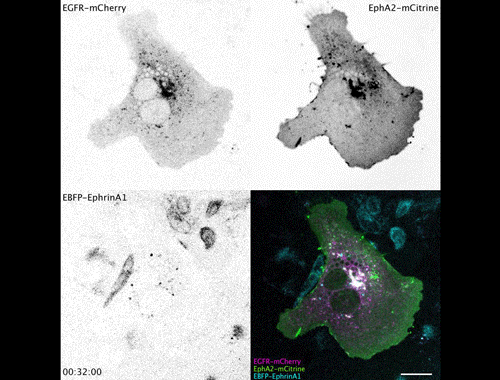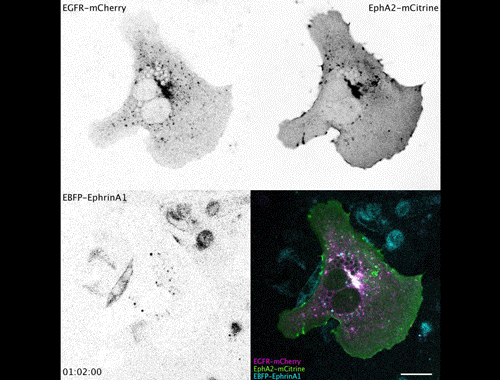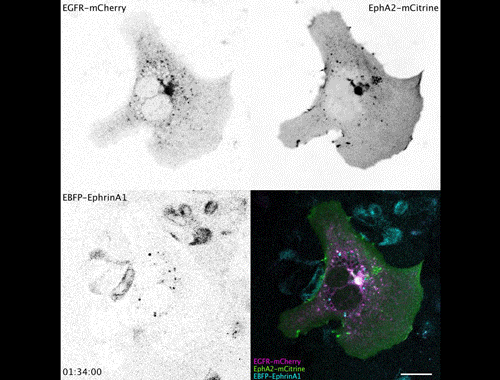
Research Assistant in Developmental and Regenerative Medicine
Department of Physiology, Anatomy and Genetics & Weatherall Institute of Molecular Medicine, University of Oxford
Grade 6: £28,098 p.a.
Applications are invited for an experienced and meticulous Research Assistant to join the Cardiovascular Development, Repair and Regeneration team working under the direct supervision of Professor Paul Riley and Dr Filipa Simoes. The project is funded by the British Heart Foundation.
The aim of the project and team is to decipher the cellular and molecular mechanisms involved in the regenerating epicardium of the adult zebrafish heart. The Research Assistant will be responsible for carrying out a range of molecular and cell biological procedures, as well as assisting with experimental zebrafish work. Previous molecular biology and embryology experience is essential but relevant training in cardiovascular development and state of the art techniques will be provided where necessary. A range of different techniques will be used including CRISPR/Cas9 technology, Nanostring analysis, in situ hybridisation and multiplex hybridisation chain reaction, tissue cryosectioning, immunofluorescence microscopy, microinjection of zebrafish embryos, cell sorting, and management of wild type and genetically modified zebrafish lines. The post is ideally suited for a candidate with an interest in developing a career that involves working at the interface of Developmental Biology and Regenerative Medicine, with a strong background in the former.
This project is a collaboration between the Departments of Physiology Anatomy and Genetics, and the Weatherall Institute of Molecular Medicine.
Candidates must have a degree in a relevant field. Expertise in molecular biology techniques, histology, and imaging, with experience of animal models including husbandry and small animal surgery is preferable.
You will be based across two sites: the Sherrington Building, South Parks Road, Oxford, OX1 3PT and the Weatherall Institute of Molecular Medicine, University of Oxford, John Radcliffe Hospital, Oxford, OX3 9DS.
The position is offered until 31 December 2018. An early start date is preferable.
The closing date for applications is 12.00 noon on Wednesday 29 November 2017. Interviews will be held on Thursday 14 December 2017.
Position Summary: A postdoctoral position is available in the laboratory of Dr. Kristin Gribble in the Josephine Bay Paul Center for Comparative Molecular Biology and Evolution, to study the role of mitochondrial homeostasis in neurodegenerative disease. This project takes a multidisciplinary approach using phenotypic, transcriptomic, genetic, genomic, epigenetic, biochemical, and imaging methods in a new animal model system, the monogonont rotifer.
The Gribble lab focuses on understanding the cellular, epigenetic, and evolutionary mechanisms of aging. We are specifically investigating the genetic and epigenetic mechanisms of maternal effects in determining offspring health and lifespan. The successful applicant will have the opportunity for independent and novel research in an innovative new laboratory. For more information about the laboratory, please see our website, http://www.mbl.edu/jbpc/gribble/ or contact Dr. Gribble at kgribble@mbl.edu.
The position is available immediately, and is renewable annually depending upon progress.
Basic Qualifications: Applicants should posses a Ph.D. and/or M.D. in molecular biology, cell biology, biochemistry, genetics, or a related field. The ideal candidate will have a record of scientific rigor, productivity, and creativity; the ability to work both independently and as part of a team; and a strong publication record. Excellent oral and written communication skills are required. Highly motivated individuals with experience in other model systems are encouraged to apply.
Preferred Qualifications: The preferred applicant will have a background in mitochondrial biology, developmental biology, and/or neurobiology. Prior experience in confocal microscopy, RNAi, and/or bioinformatics is beneficial.
Embryonic patterning is dependent on the establishment of the anteroposterior (AP) and dorsoventral axes early in development. In mammals this occurs by a breaking of symmetry in the epiblast, however the molecular mechanisms controlling this process are still not fully understood. This week we feature a paper published in the latest issue of Development that models these patterning events in gastruloids. Two authors: David Turner and Peter Baillie-Johnson from the Martinez-Arias group at the University of Cambridge told us more.
David Turner and Peter Baillie-Johnson
David and Peter, can you give us your scientific biographies and the main questions the lab is trying to answer?
DT Our lab is primarily concerned with understanding the early decisions involved during mammalian development. Specifically, how the early mouse embryo patterns itself and specifies the body axes. We mainly use mouse embryonic stem cells as a model system to study these early developmental processes both in normal monolayer culture conditions (2D) and also using our gastruloid model system in 3D (embryonic organoids).
DT For quite some time I’ve been interested in cell signalling in general and how cell fate is determined, an interest which was initially sparked during my Pharmacology honours degree in Liverpool. Following in this vein, I took up a PhD (also in Liverpool) with Prof. Mike White (who’s now in Manchester) where I used single-cell fluorescence imaging to investigate the dynamics of NF-kB in response to low doses of the cytokine TNFa.
PBJ I studied Natural Sciences as an undergraduate at Cambridge from 2010-2013, specialising in Genetics in my final year. I started my PhD in October 2013 and handed in the final copies of my thesis in October 2017. During my PhD, I worked on developing the gastruloid system and applying it to the origin of the mammalian spinal cord. Since April this year, I’ve been working as postdoctoral research assistant to Professor Jenny Nichols, who has helped me shift my focus to earlier stages of mouse development, namely gastrulation.
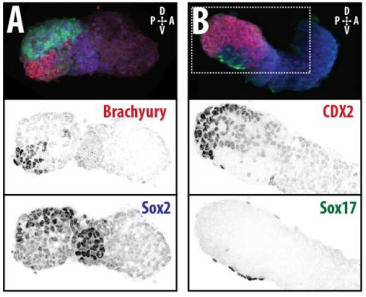
Axial organisation of gastruloids from Figure 1, Turner et al, 2017
David and Peter, how did you both come to join the Martinez Arias lab?
DT Having finished my PhD in Liverpool, I saw a post-doc position open in Alfonso’s lab which was about using mouse embryonic stem cells as an in vitro model system to try and understand their properties and their differentiation potential. My background from my PhD was strongly in single-cell imaging and cell signalling so it was a perfect opportunity for me to pursue my interests.
PBJ I first joined Alfonso’s lab in 2012 as a summer student during my undergraduate course. At that time, the lab was more focused on the regulation of embryonic stem cell pluripotency and differentiation. I was really taken with this introduction to stem cell biology and felt warmly welcomed into the lab, so I applied to follow the work up in my final year research project. While awaiting the results of my finals, I ran into David and heard about the first gastruloid experiments, which naturally became the focus of my PhD.
Your paper addresses the question of axis establishment in the early mammalian embryo. What was known about the molecular control of polarity prior to your paper?
PBJ The textbook models of antero-posterior axis specification in the mouse describe an opposing arrangement of signals and their inhibitors emanating from the extra-embryonic tissues that surround the then radially-symmetric epiblast. These models describe how the signals in the future posterior are balanced in the future anterior by their corresponding secreted inhibitors. The cells of the epiblast are then restricted to undergo a localised EMT (i.e. the beginnings of the primitive streak) only in the future posterior region, while the anterior epiblast remains reserved for the anterior nervous system. The key feature of this model is that the asymmetry originates in the extraembryonic tissues, which then becomes conferred on the underlying epiblast. Following our study, it now seems as if this careful balance of signalling across the embryo might act to permit an intrinsic symmetry-breaking event in the future posterior, rather than actively instructing the process. It remains to be seen whether the lack of the anteriorly expressed inhibitors in the gastruloids fully explains the lack of anterior structures that we have observed.
Why did you use gastruloids and not an in vivo system for this project?
PBJ I think that the acquisition of antero-posterior polarity is a good example of a topic that has been well-described in the embryo through genetic experiments. I think the strength of the gastruloids as an experimental tool is in providing insights on development that would not be possible by looking at the embryo alone – in this case by looking at development in the absence of the extraembryonic tissues and the post-implantation mechanical microenvironment. This In vitro system also enables the possibility of identifying the sufficient components behind a genetic process by starting with a minimal set of interacting parts. This approach is quite different to genetic experiments, which are good at identifying the necessary components of developmental systems, but which can’t (easily) demonstrate sufficiency.
DT Echoing what Peter said, the gastruloids have a significant advantage over in vivo systems in that the developmental processes and events we’re interested in occur at a time in the embryo that’s difficult to access and manipulate experimentally. With our gastruloids, we’re able to ask very specific questions about subtle timings of signals in ways which are either very difficult or impossible to do in the embryo, such as pulses of signals at very precise temporal intervals. Compared with in vivo models, gastruloids are relatively inexpensive, easy to manipulate, and amenable to experimental perturbation. Also, because of this ability to recapitulate many of the early developmental processes (mentioned above), our system has the real potential to be used as a way to reduce or replace animals used for research in development, which is central to the aims of NC3Rs (National Centre for the Replacement, Refinement and Reduction of Animals in Research).
Gastruloids recapitulate early embryonic events, from figure 2, Turner et al, 2017
Can you give us the key results of the paper in a paragraph?
DT The main aim of the paper was to use gastruloids to study anteroposterior patterning. We found that gastruloids spontaneously break symmetry, polarise gene expression and undergo axial elongation in a robust and reproducible manner similar to the early embryo. One end of the gastruloid resembles the posterior region of the embryo, where Brachyury is up-regulated. Our quantitative analyses show that this is regulated by both Nodal and Wnt/b-Catenin signalling, and surprisingly with no detectable involvement of BMP signalling. Our most interesting finding is that the AP axis can form without any of the extraembryonic tissues which have been suggested to be important for this process. This lead us to hypothesise that the role of the extra-embryonic tissues in the embryo may not necessarily be to induce the AP axis, but to bias the intrinsic symmetry-breaking potential of the embryo.
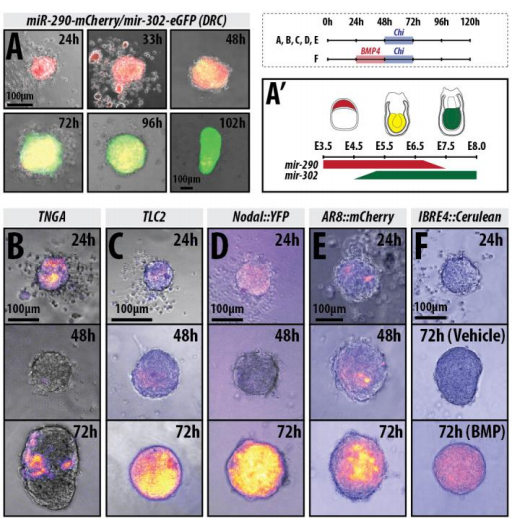
Wnt signalling enhances gastruloid polarisation, from figure 3, Turner et al, 2017
To what extent does gastruloid patterning recapitulate embryonic development?
DT I think that the gastruloids can recapitulate many aspects of early post-implantation embryonic development in the mouse and at a similar timescale to the embryo. First and foremost is their ability to develop AP polarity with Brachyury expression at one end (which we have designated the posterior) and in the DV direction where Sox2 is directly opposite Sox17. Furthermore, they’re able to extrude cells from the extending, Brachyury-positive region, in a manner akin to gastrulation at the primitive streak (hence their name). Gastruloids do not, however, develop the pre-occipital tissues of the head and brain, so their antero-posterior axis probably only represents the post-occipital levels of the embryo. We think this is due to a lack of tissues that would protect this region from high levels of ‘posteriorising’ signals (such as the prechordal plate and anterior mesoderm). It remains to be seen exactly which axial levels are represented in these seemingly posterior tissues and what we need to do to expand this representation.
PBJ I think an interesting feature of gastruloid development is the time over which the events unfold, which in our standard cultures corresponds approximately to the five days after implantation. It’s striking how they always undergo the same progression of changes in gene expression and morphology in this time and this is, for me, a key reason for using them to investigate early developmental events such as gastrulation.
In addition to your own gastruloid research, other recent papers have described systems that aim to recapitulate early development in vitro. This has generated considerable media attention and has sometimes been described as ‘creating artificial life’. How do you feel about this description? More generally, do you think there are any ethical issues thrown up by research using so-called synthetic embryos?
PBJ I think that this is a sensitive issue that certainly demands careful ethical consideration – perhaps in a longer form than an interview. Although it’s a semantic point, I think that the description of “creating artificial life” is imprecise and unhelpful and I’d prefer to see “engineering developmental systems” used instead. I think that this emphasises that these systems are controlled approaches that can recreate features of embryonic development, but which might otherwise be limited in their developmental potential.
Many of the ethical considerations surrounding these experiments and ultimately the scope of the legislation that will regulate them hinge on how exactly we define an embryo. For example, if researchers constructed a set of tissues from specific cell types in the exact image of the early embryo, would we consider the two to be equivalent? Conversely, how would a structure that closely resembles a significant proportion of the embryonic body measure up to the embryo if the representation was not complete? Would this logic extend further to individual organs or organoids? I think that scientists and the public need to consider these issues if we are to determine how we will regulate this form of research alongside the existing framework for research with embryos (which may itself need to be revised). With a clearer definition in hand, it will be easier to approach the deeper questions of where to draw appropriate limits of research on mouse and human systems, whether engineered or otherwise. To paraphrase a commentary from Martin Pera et al.1, the extent to which researchers can recreate embryogenesis in vitro will come to define not only how experimentally useful these systems are, but also how much scrutiny they will attract.
When doing the research, did you have any particular result or eureka moment that has stuck with you?
DT I think one of the best moments was late in 2013 when Alfonso and I were watching a time-lapse movie on the microscope that Susanne van den Brink had just finished imaging (the lead author on the first gastruloid paper). This is where we saw, for the first time, cells being extruded from the elongating region of the gastruloids and realised how important and useful this system was going to be. A second important moment experimentally was during the early stages of optimising the gastruloid protocol, when we were trying to improve the reproducibility within each plate and between plates of gastruloids. We found that the simple act of adding a second wash of PBS improved all the stages of gastruloid formation: the aggregation, the patterning and the elongation, so this felt like an important breakthrough.
PBJ There have been a couple of those precious moments when you’ve seen something that no-one else has before, which have been real highlights for me. I think I was lucky to have worked on such an exploratory project for my PhD as those moments have really stayed with me. In general, however, the work has progressed incrementally but I’ve been consistently surprised by the level of autonomy that the gastruloids show as we’ve started to look more closely at their development.
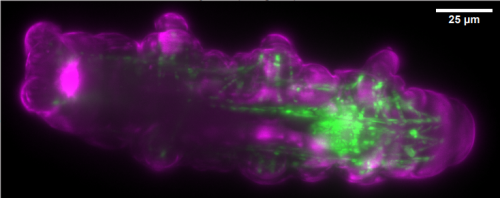
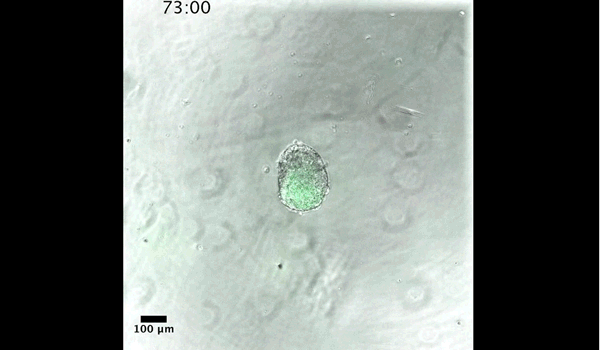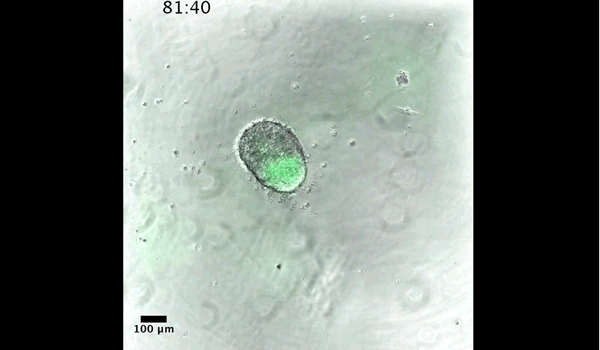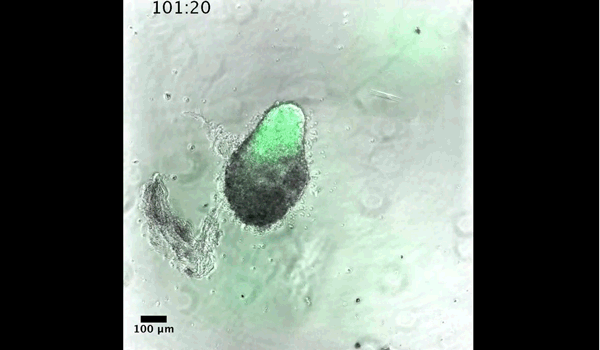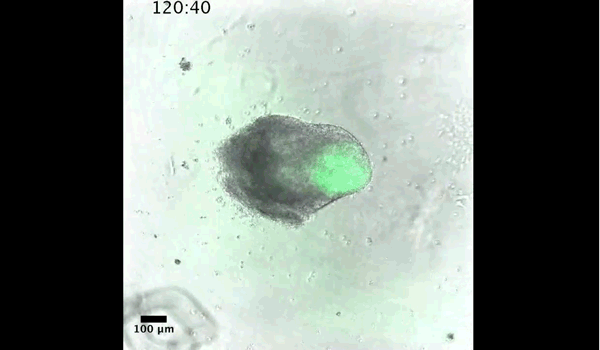
Gif generated from supplementary movie 2: Live-imaging of gastruloid development. From Turner et al, 2017
And what about the flipside: any moments of frustration or despair?
DT We found, after quite a few frustrating early attempts, that the initial culture conditions are essential to ensure good formation of gastruloids, i.e. low passage numbers, consistent splitting ratios, plating density. It took a little while for us to realise this and to ensure that our stock flasks are treated in a consistent manner.
PBJ There were definitely frustrating times in the early stages of my PhD, when we hadn’t identified the key variables that could determine whether the cultures would thrive or fail. We also had moments of doubt as to whether our observations would prove to be new and useful biological insights, but our confidence in using gastruloids as an experimental tool has grown as we’ve learned more about their development.
What are your career plans following this work?
DT I was recently awarded a David Sainsbury Fellowship from the NC3Rs to use the gastruloid system to study left-right asymmetry during mammalian development, and for the next three years I’ll be working pretty much solidly on that!
PBJ I’m currently working as a postdoctoral Research Assistant to Professor Jenny Nichols, who is helping me to cut my teeth on mouse embryology. By studying the gastrulating mouse embryo first hand, I’m trying to determine how closely the process of “gastrulation” in the gastruloids measures up to that in the embryo. By doing so, I hope to determine whether gastruloids could be used as an experimental tool to dissect this complicated phase in the life of the embryo. My work is closely aligned to that of a Cambridge-based consortium that is investigating gastrulation through single cell genomics, so I hope that my work with the embryo will provide a reference for their findings and that the gastruloid system might offer a complementary approach in the future.
And what next for the Martinez Arias lab?
DT There are quite a few avenues of our research at the moment. One is to get more of a handle on what drives the elongation in gastruloids, whether it is a mechanism based purely on convergent extension or rapid cell growth, and whether the signals suggested to be involved during the axial extension of the embryo work in a similar manner in gastruloids. We’re also interested in seeing whether the gastruloid system is applicable to later stages of development and what its limitations might be; our ongoing collaboration with our co-authors in Matthias Lutolf’s lab at EPFL is an important part of this research.
Finally, what do you two like to do when you are not in the lab?
DT As little as possible to be honest, since free weekends and evenings are quite hard to come by when stem cell work is involved! Any free time I have I like to spend with my wife and my two children. Otherwise, I spend plenty of time reading and am currently working my way through Stephen King’s Dark Tower novels, which I can certainly recommend.
PBJ I love to be outdoors, so try to get out walking, running or cycling at the weekends. I’m also a keen cook and enjoy testing out new recipes on my friends and family.
The Mammalian Genetics and Development Workshop is an annual meeting covering the development and genetics of mammals. The Meeting is based on the submitted abstracts, and include diverse topics ranging from mammalian development (not exclusively human or mouse) and identification of disease genes and mechanisms, to human genetics, epigenetics and association studies. Other model systems (including Drosophila, zebrafish and chick) are also welcomed where these relate to general developmental questions and/or disease models.
The meeting will be similar format to the 27th Meeting in 2016, which had an excellent set of 22 short talks covering various aspects of developmental biology, mouse models of human disorders, as well as talks on epigenetics and genetics of human disease (abstracts at https://doi.org/10.1017/S0016672317000039)
The workshop is traditionally a venue for post-docs and PhD students to talk rather than laboratory heads and is an excellent training ground and a friendly, informal forum. In keeping with this objective, we offer TWO PRIZES of £150 to individual post-graduate/post-doctoral presenters.
Registration
A £10 registration fee is payable by all attendees on arrival at the meeting. This fee covers the abstract booklet, tea and coffee refreshments and the wine reception. Speakers and chairpersons will be provided with lunch, free of charge, on the day of their presentation. Other participants will be expected to make their own arrangements for lunch.
Abstract Submission
All Workshop presentations will be in lecture format (15 or 20 minutes). Please send your abstract by e-mail (Word or rtf file) to ich.mgdwshop@ucl.ac.uk by 5pm on 8th November. In addition please indicate the length of talk you prefer.
Abstract format is as follows: Title (bold), AUTHORS (ALL CAPS), Address (italics) and text (200 word limit). Abstracts will be published (with the authors’ permission) in Genetics Research (https://www.cambridge.org/core/journals/genetics-research).
Established by the British Society for Developmental Biology in 2014, The Gurdon/The Company of Biologists Summer Studentship scheme provides financial support to allow highly motivated undergraduate students an opportunity to engage in practical research during their summer vacation. Each year, ten successful applicants spend eight weeks in the research laboratories of their choices, and the feedback we receive is outstanding.
Our sixth report from the 2017 group of student awardees comes from Katarina Grobicki (student at University of Cambridge), who undertook her studentship withSeb Shimeldat the Dept. of Zoology in Oxford.
Hedgehogs & Sea Squirts
Receiving a Gurdon studentship allowed me to join Seb Shimeld’s lab in for a brilliant summer in Oxford and has given me my first taste of independent research. My project focused on the hedgehog signalling pathway in Ciona intestinalis. Hedgehog signalling regulates many areas of development, including the nervous system and limb patterning, and also regulates the behaviour of adult stem cells so misregulation can lead to a variety of cancers.
In vertebrates, Hh binds to the cell surface receptor Patched, lifting the repression of Patched on Smoothened and allowing signal transduction. In cells not receiving Hh signal, Gli transcription factors are processed to a repressor form (GliR); when Hh signalling is received, this cleavage is inhibited allowing the longer activator form of Gli to persist (GliA). The balance of GliR:GliA is what determines the expression levels of target genes.
In addition to this protein level regulation, signalling also feeds back to regulate the expression of the Gli genes themselves. Ciona are of interest because they are urochordates, so are part of the invertebrate lineage most closely related to vertebrates, which split off before the 2R genome duplication. Vertebrates have 3 Gli genes and their expression is differentially regulated by Hh signalling, whereas Ciona only have 1 Gli gene. I wanted to investigate whether the Ciona Gli gene was also regulated by Hh signalling, or whether this was a novel innovation in vertebrates, made possible by the the 2R duplication. Making riboprobes
The first step of my project was to synthesise riboprobes for genes involved in the hedgehog pathway (Hh, Ptc, Gli) and a suitable control (FoxIc). Initially I attempted to do this by using PCR with M13F&R primers to amplify the target cDNA from a mini-prepped plasmid, then in vitro transcription to synthesise RNA from this. However after 6 separate PCR and transcription attempts I was still encountering problems. Every PCR of the Gli plasmid produced two DNA fragments (seen by running the products on an agarose gel), even when the primers were swapped (for T7 and T3 primers), a gradient of different temperatures were tried, new PCR reagents were used, and a fresh dilution of the plasmid mini-prep was used. This meant that the Gli PCR product couldn’t be used for probe synthesis. I had successfully amplified the other cDNAs (Ptc, Hh, FoxIc) by PCR however not enough RNA was being produced by the transcription reactions so I decided to restart the probe synthesis from scratch using a different method.
I used restriction enzymes to linearise the plasmids and then transcribed from this linear DNA to successfully produce riboprobes. However the probes were still produced in very small amounts, so I transformed competent cells with my plasmids and grew a mini-prep in order to try again with a much larger initial concentration of DNA. Using the mini-preps I produced large enough concentrations of both positive and negative sense probes for Gli and FoxIc (negative sense probes were used as controls in in situs), and positive sense probe for Ptc, and I purified these using phenol and chloroform.
It was not possible to produce as negative sense probe for Ptc due to the Ptc plasmid also containing around 200bp of cDNA encoding part of an RNA-binding protein (this is due to the way the plasmid library has been constructed and this has been missed in previous studies of Ciona Ptc expression). Any riboprobe synthesised using T3 RNA polymerase would also bind to mRNA encoding the widely expressed RNA-binding protein, rendering the probe useless. To produce the positive sense Ptc probe, EcoRI had to be used instead of Xba, so that the DNA encoding the RNA-binding protein was not included.
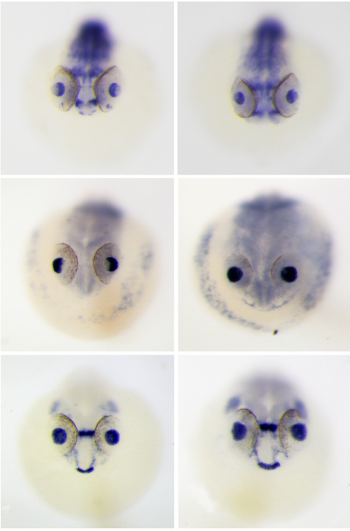
Once I had produced my probes it was time to collect some Ciona and begin in situ hybridisations. Ciona work
We drove to a harbour on the south coast to collect gravid adult Ciona which I used to set up in vitro fertilisations. Dissecting Ciona for the fertilisations was tricky and required very steady hands in order to collect eggs without cutting the sperm duct (essential to avoid self-fertilisation). I set up multiple separate fertilisations in the evenings and then incubated these at 17°C overnight, which meant embryos reached early-mid tailbud by the next morning. I then fixed the embryos in paraformaldehyde (PFA) at different ages, ready to use for in situ hybridisations. For earlier stages which had not hatched I had to dechlorionate the embryos before fixing; this must be carried out very rapidly to avoid damaging the embryos. In situ hybridisations
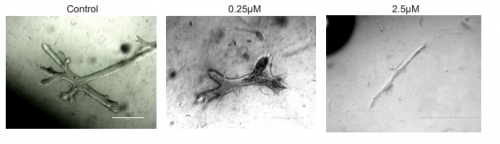
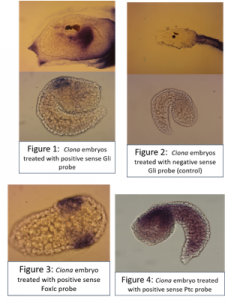
After fixation, embryos were washed in ethanol then prehybridised, before adding 3ul of probe to each eppendorf. The probe was thoroughly washed away and embryos were incubated with blocking solution, then with antibody, then washed many times again before adding the staining solution.Larvae treated with the positive sense Gli probe clearly showed real staining in the head, and tailbud embryos seemed to show staining in neural tissue along their dorsal side, but this was less clear due to background in some of the embryos. In addition, unfertilised eggs were evenly stained all over with the positive sense probe but not stained when treated with the control probe.Embryos treated with the FoxIc probe showed the predicted expression pattern, reassuring me that my protocol was working.My Ptc probe required multiple in situs to optimise the pre hybridisation procedure and find the ideal amount of probe to add. Eventually I was able to see staining towards the back of the head on dorsal side (possibly in ectoderm), but there is still quite a lot of background.Cyclopamine experiment
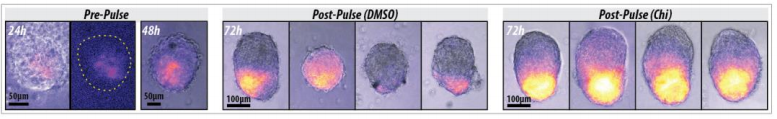
I carried out another set of fertilisations and treated the embryos with cyclopamine (or DMSO as a control) once they reached early tailbud stage; the embryos were fixed in PFA after hatching then used for in situ hybridisations. Cyclopamine inhibits the hedgehog pathway by directly binding to Smo (Chen, J; Taipale, J; Cooper, M; Beachy, P; 2002. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev, Nov 1; 16(21): 2743–2748). I wanted to test how cyclopamine would affect Gli and Ptc expression in Ciona because they only have one Gli gene, which may not be transcriptionally regulated by Hh as it is in vertebrates with their 3 Gli genes. We predicted that Gli expression would not be altered by inhibiting the hedgehog signalling, but Ptc expression would be altered. Unfortunately my embryos stained far too darkly due to me failing to fully remove their outer tunic during prehybridisation, and I ran out of time to repeat this experiment, so I wasn’t able to observe how Ptc and Gli expression patterns were affected.
A few of the cyclopamine-treated embryos with the Gli probe didn’t stain too darkly so expression patterns could still be seen, and these showed the wild-type expression pattern of Gli, which suggests that Gli is not regulated by Hh signalling. However as my Gli controls and all of my Ptc embryos were stained too darkly to see any expression patterns, I can’t conclude anything from these results, as it may have been that the cyclopamine treatment didn’t work. This experiment will hopefully be repeated in the lab soon.
Alternative splicing
I also briefly investigated whether Ciona Gli might be alternatively spliced, as in Amphioxus. To do this I extracted RNA from Ciona larvae then reverse transcribed from this using oligo(dT) primers to produce cDNA. I carried out a PCR using primers which had been designed to amplify around exons which had been identified as potentially not being included in all transcripts (using the different transcript models available on ANISEED, a very useful integrated database of ascdian genome and expression data). I then ligated my PCR product into a T vector and transformed this into competent cells, which I grew up and then sent for sequencing. My sequence data suggested that Gli isn’t alternatively spliced. My sequence data also suggests that neither the KH2012 transcript model nor the JGIv1.0 transcript model are quite correct in their predictions of exons in Gli, as my cDNA contains 2 exons not predicted by the KH2012 transcript model and doesn’t contain an extra exon predicted by the JGIv1.0 transcript model.
Thank you so much to Seb for hosting me, and to the whole lab for being so welcoming and teaching me so much. Thank you to the BSDB for making it all possible. I really feel like I’ve learned a lot over the summer and I’m now certain I want to pursue a PhD and hopefully a career in research.
Christiane Nüsslein-Volhard is Director Emeritus at the Max Planck Institute for Developmental Biology in Tübingen, Germany. In 1995, she was awarded the Nobel Prize for Physiology and Medicine, along with Eric Wieschaus and Edward Lewis, for her work on the genetic control of embryogenesis using the fruit fly Drosophila melanogaster. In the 1990s, she transitioned her lab to working with zebrafish (Danio rerio), using similar forward genetic approaches to those that had proved so successful in Drosophila to uncover key regulators of vertebrate development. We met with Christiane at the recent International Society for Developmental Biology (ISDB) meeting in Singapore, to talk about her research, the impact of the Nobel Prize and the challenges of being a ‘woman in science’.
Let’s start at the beginning: what got you interested in biology in the first place?
I was interested in animals and plants from very early on. I can’t remember having been taught it as a young child, but I was always curious, even before I went to school. I collected plants from the garden and so on, and I remember that a father of a school friend of mine collected butterflies – with which I was fascinated – and later I also joined a bird-watching club. I still love looking at nature like this – I have a big garden and I’m very interested in gardening and wild flowers. Once I got to secondary school, we had very good science teachers, and of course that helped as well.
Your undergraduate and PhD degrees were in biochemistry and molecular biology, but you’ve said that you started to get bored with that field towards the end of your PhD thesis. What attracted you to developmental genetics and how did you transition into that field?
I actually started my undergraduate studies with biology and I found the lectures rather boring, so I switched to physics, which at the time was fascinating and was taught by a very good professor. I also really enjoyed physical chemistry, and I moved into biochemistry – in which I eventually got my diploma – in order to get a solid background in basic science.
My PhD was in molecular biology, but I’m not a molecular biologist or a biochemist. I went into this area because I was ambitious and at that time it was the field that was really moving forwards and was fashionable. But at the end I found I wanted to do more organismal biology. I think I went into developmental biology at least partly because Alfred Gierer’s group in our institute was working with Hydra, trying to isolate the factors that would specify ‘head’ and so on. Being exposed to this work, I became interested in the question of morphogens. The Gierer-Meinhardt model on morphogen gradients had just been published, and though I didn’t fully understand it, I could see that morphogens were becoming a big issue in the field. However, people were against gradients because they hadn’t been identified.
I thought that to identify morphogens you had to combine genetics and developmental biology: if you could make a mutation in a gene that encoded a gradient and see the consequences of its loss, you would be on the way to finding them. I wrote a proposal for an EMBO fellowship that spelt this idea out, and this was how I ended up in Walter Gehring’s lab working with Drosophila. Gehring kept the strain of the only potential morphogen mutant (identified by Alice Bull in 1966) at the time, which had mirror image duplications of the posterior end that no one understood, although Meinhardt explained to me that it must be a gradient mutant. This was bicaudal, and I tried to understand it but it was the most difficult mutant I ever worked on. So instead I developed methods to make my own mutants and finally ended up at EMBL with Eric Wieschaus, where we started the large-scale screens.
And this is probably the work you’re best known for and that won you and Eric the Nobel Prize – the large-scale genetic screens in Drosophila embryos. Did you think in advance that this work would be so influential?
Looking back, it is amazing how little was known about anything at the time. For example, no one knew anything about how segmentation could work, or about polarity – at least beyond the theoretical work. So it was a completely open area and whatever you touched would turn out to be interesting. For example, in the first half-year in Walter Gehring’s lab I isolated the dorsal mutant, which is a gradient mutant. Lucky me – that was exactly what I’d been looking for! And then we started to find these segmentation mutants – they just popped up when you looked at large numbers of embryos. But strangely enough Eric and I seemed to be the only people who saw the potential in screening for mutants. Somehow, we were far ahead of everyone else – who all thought that screens were too much effort and that they might not work, or that you’d only find ʻmessy’ things.
As I’ve said before, there was also this idea that there were no gradients; many people at the time believed in specific localised determinants rather than gradients. Some groups that did do screens looking for particular kinds of mutants (maternal homeotic mutants) failed completely because they couldn’t interpret the mutants they found – they didn’t fit with their model for how development would work. So when we did our screen, we found things that people had isolated before but hadn’t recognised for what they were.
When we started, we had different research backgrounds and our own interests – I was pushing to do maternal screens to try and get at the morphogens, which was genetically very tricky, while Eric was particularly interested in oogenesis and sex determination. But we had a very small lab with just one technician between us and so we realised it would be better to do things together. So we decided to do the large-scale zygotic screens together, and we were amazingly successful. We were a good team: we had similar observational qualities and were both pushing hard. Three years of collaboration was enough – we were both strong characters – but it worked incredibly well for that time.
One big question was: now we understand something about fly embryogenesis, what about vertebrates? Do they do things the same or completely differently?
And what came out of the screen? Did you uncover genes that play key roles across evolution and did this in some way trigger your starting to work with zebrafish?
Actually, the highlights in the fly screens aren’t conserved at all. In the first evaluation we focussed on segmentation because this is easiest – you can cleanly count the segments and look at polarity and so on. This gave us the pair-rule genes and the gap genes, which were exciting because they told us a lot about an important developmental hierarchy, but these principles aren’t conserved beyond insects. And then when I moved to Tübingen, we did the maternal screens to get the morphogens (which I wanted from the beginning) – we found genes such as bicoid and oskar, but again these are not conserved.
So, one big question was: now we understand something about fly embryogenesis, what about vertebrates? Do they do things the same or completely differently? At the time, the people working with frogs (which were the main system for vertebrate developmental biology back then; the mouse was not that well developed) were a completely separate community from the fly people, and they thought that the fly couldn’t tell them anything. The prevailing view was to explain everything with ‘factors’, and to approach developmental biology through embryological rather than genetic manipulation, while we explained everything with genes and genetics. And I was convinced that the genetic approach was the better one and that you needed to be able to do genetics in vertebrates.
It was only around that time, beginning with the homeobox genes, that the homology between invertebrates and vertebrates slowly became apparent. But we had decided to start a screen in zebrafish before we fully appreciated the degree of conservation, and actually the screen turned up many of the same genes that had already been identified as key factors in frog, so we were perhaps a bit late with this. The biggest question in vertebrate development at the time was how gastrulation works and how the axes are organised, and Eddy De Robertis, Marc Kirschner, Jim Smith and others had already identified things like the critical BMP gradients, so when we identified and cloned the equivalent fish mutants they weren’t a huge surprise.
How difficult was it to transition your lab – which had had such success working with flies – to zebrafish?
Well, by then the fly lab was not that big anymore. My lab style has always been that postdocs took their projects with them, and some of the graduate students too: Ruth Lehmann took oskar and a bunch of mutants with her, and Kathryn Anderson took most of the dorsal group genes. So people had gone away with some of the most interesting projects. When the screens were finished, we began to clone some mutants, which I found tedious and did not like too much, so I decided I wanted to move to fish. However, then the Bicoid gradient was discovered by Hans Georg Frohnhöfer and Wolfgang Driever and the Dorsal gradient by Siegfried Roth (all graduate students in the lab), and we were working very successfully with these projects and the transcription factor hierarchies, and doing lots of biochemistry, and this kept us busy for quite some time. Two graduate students, Stefan Schulte-Merker and Matthias Hammerschmidt, started working with fish on the side during this period, and when the fish screen really got started the fly lab was gradually coming to a natural end.
In 1996, Development published a Special Issue devoted to zebrafish, including the Nüsslein-Volhard lab screens. Christiane was co-author on 25 of the issue’s papers.
Setting up a screen in a new organism is a lot of work, but I like this kind of work – developing devices and tools that make things easier and more efficient. This was also the secret of success in the fly screen. It’s maybe my upbringing from home – we used to do handicrafts and make toys and I had quite some dexterity in this sense. Eric is also good at these things, so we invented a bunch of tools and tricks that made life much easier – little things like boxes with a grid for keeping fly stocks (rather than just holding them together with rubber bands as everyone did at the time), and a better system for collecting embryos on agar plates instead having to take them out of the food. And we found a particular type of oil that made embryos transparent so you could look through the chorion, which just made things so much easier and faster for screening.
With the fish it was a similar situation: we needed to be able to make all the processes as efficient as possible to do a large-scale screen. I went to Oregon, where zebrafish were quite well established as a model system, but realised that we couldn’t possibly do a screen with the way they raised fish – it was too laborious. So the first job – and it took two years or so – was to develop better aquarium and feeding systems and so on to streamline everything and allow us to scale up.
Your work now focusses on pigment patterning in adult zebrafish, and in your talk here you introduced the topic as being about the ‘evolution of beauty’, discussing Darwin’s interest in this problem. Did these aesthetic and historic aspects draw you to the problem?
I think the Darwin link came later to be honest. Initially, I was just struck by the beauty of the fish, like I had been by the segmentation pattern of flies: it’s always nicer to work on something you find beautiful. Working with fish, it’s natural to wonder how the pattern is made, and we found a number of mutants with aberrant pigment patterns and started to work on those. To some extent, talking about it in terms of beauty is advertising – it’s a way of explaining why you’re working on this obscure problem. And the more you talk about it the more you think about it, and realise that the stripes must have a function – they must be important. Then I read Darwin’s Descent of Man, which is a fascinating book. He thought about the issue of why humans look different across the world – skin colour, hair colour and so on. At Darwin’s time, some people still thought that black and white people were different species (with the whites superior), and Darwin wondered about this – why are they different, and are they really separate species? He thought that they were too similar to be different species, and he argued that they look different because they have different standards of beauty. He even collected observations from travellers to look for physiological differences (like whether skin colour had a significant effect on heat resistance), but there was no really good biological explanation for why they looked different. So he came up with the idea that it was to do with attractiveness. And when you look around the animal kingdom you see the same thing. The book is partly about sexual dimorphism in the animal kingdom, and beauty is of course very important. When you look at animals like peacocks or birds of paradise, some evolutionary biologists have argued that they grow such extraordinary feathers to prove that they’re strong enough to carry them around, or whatever, and of course that’s rubbish – it’s about beauty. I found this all very interesting, and now use it as a way of explaining why we’re interested in stripes in zebrafish when writing reviews and giving talks.
You’re here in Singapore at the ISDB meeting giving one of the Nobel Prize lectures. You’ve said in the past that winning the Nobel Prize, while a great honour, was a distraction for you scientifically. But it must have given you opportunities you wouldn’t otherwise have had. How do you feel about the prize now?
Yes, I have mixed feelings. When I first won the prize I got carried away – I got invited to give many lectures, some of which I should have said ‘no’ to but didn’t, and in the end I felt sucked dry. Then I was so exhausted that I said ‘no’ quite often, but that was also bad because it spoiled my reputation to some extent. It was just too much and it was hard to focus and to get the balance right. And in the lab, although I generally have very smart students and postdocs who work quite independently, I don’t think I was there enough to give them proper feedback and advice.
You also meet a lot of envy from colleagues, and this is painful sometimes. It has seemed to me that men can have a hard time accepting that women can be smarter, particularly when a woman points out mistakes that the man has made. Throughout my career, I often had difficulties in getting a point across because the men couldn’t bear it that I might ‘win’, and in some cases this got even more difficult after the Nobel Prize.
On the other hand, there were interesting opportunities. For example, I was elected onto the National Ethics Council of Germany. I had to learn a huge amount for this – for example, about mammalian embryology and stem cells – and this took a lot of time. It was very interesting but, to be honest, it was not fun. There was so much dispute about how to handle some of these issues, and many people were very old-fashioned and even hostile in some cases. And I lost most of the fights – I was on the liberal side and unfortunately the conservative side usually won. But it was a very interesting experience and I wouldn’t have missed it. And of course you also have the opportunity to get in contact with really interesting people through membership of learned societies and academies, and this has been a real reward.
You must have been one of only a very few women around when you started in research, and you’ve talked about how difficult you found this. How do you think the situation for women has changed over the years?
It’s much better now, though I think we’re overshooting a bit in some areas – search committees run after women in some cases, and that’s another danger – but still have discrimination in other situations. So things aren’t yet where they should be.
But, more broadly, science has also changed: it’s more competitive now with many more scientists and more bad science than there used to be, and it’s difficult to survive with an honest and ethical approach to science. I don’t think we’re in a good place at the moment: it’s so hard for people to find jobs, and most scientists don’t have permanent contracts. This means that people are forced to advertise and even exaggerate the impact of their work, and waste so much time applying for money – the funding rate is so low. I didn’t have an easy time either when I was struggling to get a job in the early days – what Eric and I were doing was seen as quite obscure – but it’s so much more difficult today. I find it quite ambiguous to talk people into doing science nowadays because I don’t necessarily see it as a very nice or easy job.
The important thing with the CNV Foundation is to teach women that it’s okay to let people help you because you can’t do everything yourself
In 2004 you set up the CNV-Stiftung, a foundation that supports women in science with children. What drove you to set up the foundation, and what has it achieved?
I have been on committees about women in science since before I was appointed as a Max Planck Director, and after my appointment I was just one of two women directors in the whole society. As I’ve said, it was very difficult at the time and I felt very awkward. People come up with all sorts of ideas to encourage women in science, and I thought that money is perhaps something that would really help. Women usually do the bulk of the housework and they often don’t think it’s okay to ask for help from outside – it took me years to decide to get a cleaner. In general, I find that men either get this stuff done by their wives or they hire someone, but women are more reluctant. So I thought it would be good to give women money to allow them to have an easier life around the house. The important thing with the CNV Foundation is to teach women that it’s okay to let people help you because you can’t do everything yourself and you need to have some free time. We support around 15 women working in Germany per year: we started out just funding students with children, because they really don’t have the money, but now we also support postdocs. But the problem of women in science is very complicated, and while I think our foundation helps, it clearly doesn’t solve things.
Do you have any advice for young scientists?
You really need to be genuinely interested in science and enjoy making discoveries. The job depends on achievements: making discoveries and publishing them. This means hard work, and if you are not rewarded by success it can be very frustrating. You should, as far as possible, avoid mainstream areas and change fields after your PhD in order to be able to develop an independent profile and work on an original, self-selected topic. Don’t listen too much to mentors and teachers: they may not be honest but just polite. So it is important to self-critically assess your own abilities in comparison with others. It is your own responsibility.
Finally, what might readers of Development be surprised to find out about you?
I wrote a cook book, in German, which was published in 2006. I love to cook, and I think there are similarities with working in a lab: the organisation, the working with your hands and so on. I didn’t do so much cooking when I actually worked in the lab (though I used to take a cake to the lab every weekend), but when I stopped doing things with my hands at work I started cooking and baking again, and I wrote this book. It is still on the market!
Christiane’s cookbook
And I love music – I play the flute and sing. I have taken lessons for the last ten years and love to sing German Kunstlieder, and I sometimes give concerts to my friends.
The research group of Dr. Elvira Mass is looking to recruit a highly motivated PhD student to join her group Developmental Biology of the Innate Immune System.
Our research focuses on tissue-resident macrophages and their homeostatic functions. Specifically, we are interested how macrophages impact organ development and function during embryogenesis and postnatal stages on the cellular and molecular level. Based on new developmental and mechanistic knowledge, we seek to improve our understanding of disease pathophysiology and treatment.
We are part of the Life & Medical Science (LIMES) Institute, which is supported by DFG and Cluster of Excellence funding and which offers a great scientific environment for young researchers.
Requirements:
Previous experience in cell biology/immunology/hematopoiesis/animal models
The willingness to work with mice is a prerequisite
basic immunological and histological techniques such as flow cytometry and immunofluorescent stainings are beneficial
Programming skills (R/Python) for transcriptome analysis are advantageous
Your application should include:
CV
brief summary of your previous work experience (max. 1/2 page)
contact information of referees and dates when you wish to start the position
optional: 1 page research proposal for your PhD project, which builds on the study Mass et al. Science 2016
We are looking for someone working on Models of Animal Regeneration to join the CRTD Dresden as soon as possible!
Your permanent position will come with a generous package, full facility support, and the lively environment of a growing, interdisciplinary campus with tight links to the medical clinics.
Our latest monthly trawl for developmental biology (and other cool) preprints. Let us know if we missed anything.
October was a monster month for preprinting (a burst of post-summer productivity?), notable for the number of preprints covering plant development, disease modelling, modelling modelling, neurodevelopment, and organisms ranging from polychaete worms to hemipterans, Arctic charr to roses (plus all the usual suspects).
The preprints were hosted on bioRxiv, PeerJ, andarXiv. Use these links to get to the section you want:
Transcriptional regulation by NR5A2 couples cell differentiation and inflammation in the pancreas. Isidoro Cobo, Paola Martinelli, Marta Flandez, Latifa Bakiri, Mingfeng Zhang, Enrique Carrillo-de-Santa-Pau, Jinping Jia, Liv Thommesen, Torunn Bruland, Natalia del Pozo, Sara Olson, Jill Smith, William R. Bamlet, Gloria M. Petersen, Nuria Malats, Laufey Amundadottir, Erwin Wagner, Francisco X. Real
The Transcriptional Logic of Mammalian Neuronal Diversity. Ken Sugino, Erin Clark, Anton Schulmann, Yasuyuki Shima, Lihua Wang, David L. Hunt, Bryan M. Hooks, Dimitri Trankner, Jayaram Chandrashekar, Serge Picard, Andrew Lemire, Nelson Spruston, Adam Hantman, Sacha B. Nelson
RNA velocity in single cells. Gioele La Manno, Ruslan Soldatov, Hannah Hochgerner, Amit Zeisel, Viktor Petukhov, Maria Kastriti, Peter Lonnerberg, Alessandro Furlan, Jean Fan, Zehua Liu, David van Bruggen, Jimin Guo, Erik Sundstrom, Goncalo Castelo-Branco, Igor Adameyko, Sten Linnarsson, Peter Kharchenko
Autosomal recessive coding variants explain only a small proportion of undiagnosed developmental disorders in the British Isles.Hilary C. Martin, Wendy D. Jones, James Stephenson, Juliet Handsaker, Giuseppe Gallone, Jeremy F. McRae, Elena Prigmore, Patrick Short, Mari Niemi, Joanna Kaplanis, Elizabeth Radford, Nadia Akawi, Meena Balasubramanian, John Dean, Rachel Horton, Alice Hulbert, Diana S. Johnson, Katie Johnson, Dhavendra Kumar, Sally Ann Lynch, Sarju G. Mehta, Jenny Morton, Michael J. Parker, Miranda Splitt, Peter D. Turnpenny, Pradeep C. Vasudevan, Michael Wright, Caroline F. Wright, David R. FitzPatrick, Helen V. Firth, Matthew E. Hurles, Jeffrey C. Barrett
Identification, Isolation, and Characterization of Human LGR5-positive Colon Adenoma Cells. Michael K Dame, Durga Attili, Shannon D McClintock, Priya H Dedhia, Peter Ouilette, Olaf Hardt, Alana M Chin, Xiang Xue, Julie Laliberte, Erica L Katz, Gina M Newsome, David Hill, Alyssa Miller, Yu-Hwai Tsai, David Agorku, Christopher H Altheim, Andreas Bosio, Becky Simon, Linda C Samuelson, Jay A Stoerker, Henry D Appelman, James Varani, Max S Wicha, Dean E Brenner, Yatrik M Shah, Jason R Spence, Justin A Colacino
The Polycomb-dependent epigenome controls β-cell dysfunction, dedifferentiation and diabetes. Tess Tsai-Hsiu Lu, Steffen Heyne, Erez Dror, Eduard Casas, Laura Leonhardt, Thorina Boenke, Chih-Hsiang Yang, Sagar, Laura Arrigoni, Kevin Dalgaard, Raffaele Teperino, Lennart Enders, Madhan Selvaraj, Marius Ruf, Sunil Jayaramaiah Raja, Huafeng Xie, Ulrike Boenisch, Stuart H. Orkin, Francis Lynn, Brad G. Hoffman, Dominic Grün, Tanya Vavouri, Adelheid Lempradl, Andrew Pospisilik
WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. Yeon-Joo Kim, Daniel Osborn, Ji-Young Lee, Masatake Araki, Kimi Araki, Timothy Mohun, Johanna E M Kansakoski, Nina Brandstack, Hyun-Taek Kim, Francesc Miralles, Cheol-Hee Kim, Nigel A Brown, Hyung-Goo Kim, Juan Pedro Martinez-Barbera, Paris Ataliotis, Taneli Raivio, Lawrence C Layman, Soo-Hyun Kim
Molecular evolutionary trends and feeding ecology diversification in the Hemiptera, anchored by the milkweed bug genome. Kristen A. Panfilio, Iris M. Vargas Jentzsch, Joshua B. Benoit, Deniz Erezyilmaz, Yuichiro Suzuki, Stefano Colella, Hugh M. Robertson, Monica F. Poelchau, Robert M. Waterhouse, Panagiotis Ioannidis, Matthew T. Weirauch, Daniel S.T. Hughes, Shwetha C. Murali, John H. Werren, Chris G.C. Jacobs, Elizabeth J. Duncan, David Armisén, Barbara M.I. Vreede, Patrice Baa-Puyoulet, Chloé S. Berger, Chun-che Chang, Hsu Chao, Mei-Ju M. Chen, Yen-Ta Chen, Christopher P. Childers, Ariel D. Chipman, Andrew G. Cridge, Antonin J.J. Crumière, Peter K. Dearden, Elise M. Didion, Huyen Dinh, harshavardhan doddapaneni, Amanda Dolan, Shannon Dugan-Perez, Cassandra G. Extavour, Gérard Febvay, Markus Friedrich, Neta Ginzburg, Yi Han, Peter Heger, Thorsten Horn, Yi-min Hsiao, Emily C. Jennings, J. Spencer Johnston, Tamsin E. Jones, Jeffery W. Jones, Abderrahman Khila, Stefan Koelzer, Viera Kovacova, Megan Leask, Sandra L. Lee, Chien-Yueh Lee, Mackenzie R. Lovegrove, Hsiao-Ling Lu, Yong Lu, Patricia J. Moore, Monica C. Munoz-Torres, Donna M. Muzny, Subba R. Palli, Nicolas Parisot, Leslie Pick, Megan Porter, Jiaxin Qu, Peter N. Refki, Rose Richter, Rolando Rivera-Pomar, Andrew J. Rosendale, Siegfried Roth, Lena Sachs, M. Emília Santos, Jan Seibert, Essia Sghaier, Jayendra N. Shukla, Richard J. Stancliffe, Olivia Tidswell, Lucila Traverso, Maurijn van der Zee, Séverine Viala, Kim C. Worley, Evgeny M. Zdobnov, Richard A. Gibbs, Stephen Richards
Mechanochemical feedback and control of endocytosis and membrane tension. Joseph Jose Thottacherry, Anita Joanna Kosmalska, Alberto Elosegui-Artola, Susav Pradhan, Sumit Sharma, Parvinder P. Singh, Marta C. Guadamillas, Natasha Chaudhary, Ram Vishwakarma, Xavier Trepat, Miguel A. del Pozo, Robert G. Parton, Pramod Pullarkat, Pere Roca-Cusachs, Satyajit Mayor
Summarizing Performance for Genome Scale Measurement of miRNA: Reference Samples and Metrics. P. Scott Pine, Steven P. Lund, Jerod R. Parsons, Lindsay K. Vang, Ashish A. Mahabal, Luca Cinquini, Sean C. Kelly, Heather Kincaid, Daniel J. Crichton, Avrum Spira, Gang Liu, Adam C. Gower, Harvey I. Pass, Chandra Goparaju, Steven M. Dubinett, Kostyantyn Krysan, Sanford A. Stass, Debra Kukuruga, Kendall Van Keuren-Jensen, Amanda Courtright-Lim, Karol L. Thompson, Barry A. Rosenzweig, Lynn Sorbara, Sudhir Srivastava, Marc L. Salit
ACEseq – allele specific copy number estimation from whole genome sequencing. Kortine Kleinheinz, Isabell Bludau, Daniel Huebschmann, Michael Heinold, Philip Kensche, Zuguang Gu, Cristina Lopez, Michael Hummel, Wolfram Klapper, Peter Moeller, Inga Vater, Rabea Wagener, ICGC MMML-Seq project, Benedikt Brors, Reiner Siebert, Roland Eils, Matthias Schlesner
HiGlass: Web-based Visual Exploration and Analysis of Genome Interaction Maps. Peter Kerpedjiev, Nezar Abdennur, Fritz Lekschas, Chuck McCallum, Kasper Dinkla, Hendrik Strobelt, Jacob M. Luber, Scott B. Ouellette, Alaleh Azhir, Nikhil Kumar, Jeewon Hwang, Soohyun Lee, Burak H. Alver, Hanspeter Pfister, Leonid A. Mirny, Peter J. Park, Nils Gehlenborg
Barriers to Integration of Bioinformatics into Undergraduate Life Sciences Education. Jason Williams, Jennifer Drew, Sebastian Galindo-Gonzalez, Srebrenka Robic, Elizabeth Dinsdale, William Morgan, Eric Triplett, James Burnette, Sam Donovan, Sarah Elgin, Edison Fowlks, Anya Goodman, Neal Grandgenett, Carlos Goller, Charles Hauser, John R. Jungck, Jeffrey Newman, William Pearson, Elizabeth Ryder, Melissa Wilson Sayres, Michael Sierk, Todd Smith, Rafael Tosado-Acevedo, William Tapprich, Tammy Tobin, Arlin Toro-Martínez, Lonnie Welch, Robin Wright, David Ebenbach, Mindy McWilliams, Anne Rosenwald, Mark Pauley
A Data Citation Roadmap for Scholarly Data Repositories. Martin Fenner, Mercè Crosas, Jeffrey Grethe, David Kennedy, Henning Hermjakob, Philippe Rocca-Serra, Gustavo Durand, Robin Berjon, Sebastian Karcher, Maryann Martone, Timothy Clark
The scent of the fly. Paul G. Becher, Sebastien Lebreton, Erika A. Wallin, Erik Hedenstrom, Felipe Borrero-Echeverry, Marie Bengtsson, Volker Jorger, Peter Witzgall
Established by the British Society for Developmental Biology in 2014, The Gurdon/The Company of Biologists Summer Studentship scheme provides financial support to allow highly motivated undergraduate students an opportunity to engage in practical research during their summer vacation. Each year, ten successful applicants spend eight weeks in the research laboratories of their choices, and the feedback we receive is outstanding.
Our fifth report from the 2017 group of student awardees comes from Jake Cornwall Scoones(student at University of Cambridge), who undertook his studentship with Anna Philpottat the Dept. of Oncology in Cambridge.
Transdifferentiation of pancreatic organoids
This summer, I had the amazing opportunity of undertaking a Gurdon Summer Studentship, working in Prof. Anna Philpott’s lab, under the guidance of Dr. Roberta Azzarelli at the Cambridge Oncology Department. Having completed first-year Natural Sciences, I was intrigued to learn more about the molecular mechanisms that underpin development. Focused on a potential transdifferentiation pathway in pancreatic cells, the project has been both fascinating and informative, teaching me many invaluable skills, from tissue culture, to growing organoids, to having the patience required for many of the complex procedures involved.
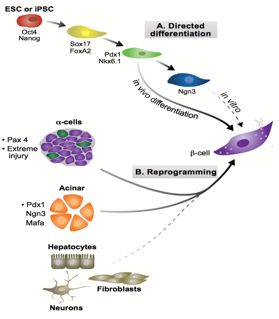
In recent years, many have championed regenerative medicine as a solution to diseases associated with cell loss like diabetes mellitus. This technique, where cells are grown and differentiated to replace dead or diseased cells, has the potential to overcome many of the pitfalls of transplantation, most notably immune rejection. To minimise rejection, inserted cells must display high genomic identity with the recipient. These cells can be derived by one of two methods:1, 2 through a differentiation programme, transitioning embryonic(-like)3 stem cells into the fate desired; or via transdifferentiation, through introducing factors that tweak the cell’s genome and epigenome.
Figure 1: Two possible pathways for generating β-cells. Adapted from Pagliuca and Melton, 2013
During development, cell-lineages sequentially accrue epigenetic modifications that consign them to their fate, transforming from a totipotent zygote to terminally differentiated cells. Waddington’s visual metaphor, envisaging this sequential differentiation as a ball rolling down a terrain of bifurcating valleys, helps to frame questions in developmental biology.4 Upon transplanting the nucleus of somatic Xenopus cells into enucleated eggs, Gurdon found that the resultant cell was pluripotent, capable of differentiating into all cell types.5 In so doing, Gurdon demonstrated that factors within an egg have the capacity to modify the epigenetic state of a nucleus, implying that the ball can roll back up the valley. In the case of pancreatic transdifferentiation, with sufficient knowledge of this terrain and with the correct molecular intervention, we should able to push the ball into our chosen valley.
Pancreatic development proceeds through two stages: the primary transition (embryonic day 9 (E9) to E12.5); and the secondary transition (E12 to birth).6 The primary transition sees endodermal thickening, followed by the pancreatic progenitor proliferation leading to pancreatic bud formation. Through a process of epithelial stratification and micro-lumen formation, the initial tubular morphology that will later characterise the pancreas begins to emerge. This initial expansion phase is followed by rounds of specification and patterning, forming a bipotent trunk and a multipotent tip. Adult pancreata are comprised of three different cell types, namely acinar, duct and endocrine cells, the former derived from tip progenitors and the latter two from trunk cells.
Endocrine development, part of the secondary transition, is triggered by the transient expression Neurogenin 3 (Ngn3) above a threshold level, and proceeds through delamination from the epithelium and aggregation into Islets of Langerhans.Ngn3+ cells are specified to one of five fates in a temporally regulated manner:7 α-cells produced upon earliest Ngn3-activation synthesising glucagon; followed by β-cells producing insulin; then δ-cells producing somatostatin; then PP-cells producing pancreatic polypeptide; and finally ε-cells producing ghrelin. Previous work from the lab by Azzarelli et al.8 demonstrated Ngn3 is phosphoregulated by proline-directed kinases (e.g. CDKs), regulating the balance between proliferation, (Ngn3 hyper-phosphorylation), and differentiation, (Ngn3 under-phosphorylation). β-cell maturation is promoted by the transcriptional regulator Pdx1, among others, whose expression remains confined to β-cells in the adult pancreas. Working in tandem with Pdx1, MafA transcriptionally regulates genes involved in insulin secretion and biosynthesis and it is thus considered a marker of mature β-cell identity.
Ngn3, crucial in assigning endocrine fate initially, and MafA and Pdx1, important in β-cell maturation, have been previously over-expressed in combination to induce transdifferentiation of acinar cells to β-fate in an in vivo model. These factors were used in our experiment to see if such transdifferentiation is possible in an in vitro model, specifically pancreatic organoids, a form of 3D cell culture grown in matrigel. Upon over-expression of these three factors, it is possible to test for changes in cell fate, by looking at symptomatic traits of β-cells, namely the production of insulin.
Over-expression can be achieved through the infection of organoids with lentiviral vectors carrying genetic loci that upon infection will be inserted into the cells’ nuclear genomes. The activation of these imported genes should be controllable so a transactivator is used which, when bound to the antibiotic doxycycline (Dox), becomes active, binding the promoter of our genes of interest, inducing their transcription. The three factors are necessarily expressed together through their transcription within a single ORF, containing MafA, Pdx, Ngn3 and GFP (a fluorescent marker) sequences, separated by loci encoding the cleavage peptide 2A, meaning upon translation, cleavage ensues resulting in four functional proteins. Hence a GFP signal is sufficient to determine the expression of all three factors, leaving other wavelengths free to test for pancreatic hormones through immunohistochemistry.
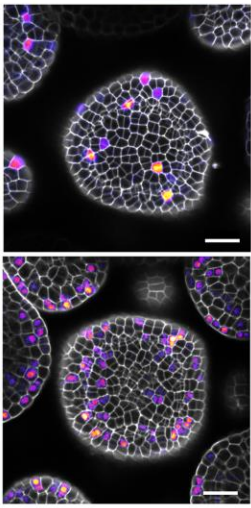
Figure 2: Pancreatic orgnaoids infected with a virus encoding GFP
Two lentiviral vectors were used to infect organoids, one carrying the transactivator gene Tet3G, and the other carrying the locus of interest, Pdx-MafA-Ngn3-GFP (PMN), or GFP as our control. Viruses were produced by the transfection of HEK cells with several plasmids, each containing genes encoding different viral proteins plus the locus of interest, followed by incubation, viral purification and titration. Organoids were incubated with viruses for a week with Dox and then fixed with PFA. Immunohistochemistry was performed, staining organoids for insulin. Preliminary results are very encouraging and I look forward to seeing how my contribution fits into the larger project as a whole.
This studentship has been an invaluable experience, allowing me to gain an understanding of real-world lab science, a practice venturing into the unknown, far from the rushed experiments for which answers were already known performed as a part of my course. I have developed lab-skills, most notably having the rare opportunity to work with organoids, a relatively novel technology. I urge other students to consider applying for this fantastic opportunity in future years.
 (No Ratings Yet)
(No Ratings Yet)
 (2 votes)
(2 votes)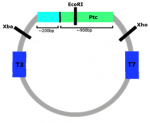
 Cyclopamine experiment
Cyclopamine experiment