We are pleased to announce that the Physics of Living Matter conference is back in Cambridge for its 19th edition!
This will be on the 24th and 25th of September 2026, at the Centre for Mathematical Sciences (Wilberforce Rd, Cambridge CB3 0WA, UK).
As per tradition, the conference will showcase a diverse set of biological problems that are tackled through the lens of the physical sciences and in addition to an exciting programme presented by renowned international speakers, oral presentations will be selected from the submitted abstracts.
The PLM series started 19 years ago from an interest in promoting the interface between the Life and Physical Sciences in Cambridge. Over the years, PLM has grown from a local meeting to a popular international event that attracts interdisciplinary scientists from all around the world.
Confirmed speakers for this edition are:
Nancy Kleckner (Harvard University, USA) – Bragg Lecture
Rosana Collepardo (University of Cambridge, UK)
William Durham (University of Sheffield, UK)
Zena Hadjivasiliou (Francis Crick Institute, UK)
Pulin Li (Whitehead Institute – MIT, USA)
Jean-Leon Maitre (Institute Curie, FR)
Jeremie Pallaci (Institute of Science and Technology Austria, AT)
Marco Polin (IMEDEA UIB-CSIC, ES)
Victror Sourjik (Max Planck Institute for Terrestrial Microbiology, DE)
Peter Swain (University of Edinburgh, UK)
Berta Verd (University of Oxford, UK)
Andrea Weisse (University of Edinburgh, UK)
The call for abstracts is open now! You can submit an abstract for a talk or a poster presentation using this form. The deadline for abstracts is 12 of June.
We will be opening the egistrations in the next few days; if you are interested, please fill in this form for updates.
For further information, you may check our website, or contact Maria Bargués-Ribera at admin@physbiol.cam.ac.uk.
We are looking forward to welcoming you to PLM19!
Best regards,
The organising committee
James Locke (Sainsbury Lab, University of Cambridge, UK) Teuta Pilizota (Department of Physics, University of Cambridge, UK) Ben Steventon (Department of Genetics, University of Cambridge, UK)
We are delighted to launch our new webinar series, Macro to micro: quantitative plant imaging across scales, with Alex Johnson and Joe McKenna. In this series, we’ll be highlighting the latest research using imaging to investigate questions in plant biology. We’ll also hear talks from imaging experts from outside the plant biology field.
You can find more information on the webinar series, including how to register for event notifications and to sign up to give a talk, on our dedicated webpage.
Our first webinar is on Thursday 30 April at 15:00 BST and will feature talks from Beatrice Lace and Simon Gilroy. Beatrice will present, ‘Multiplexing and Endogenous Fluorescence Discrimination Using FLIM in Plants’ and Simon will ask, ‘Do plants feel pain? How imaging has brought plant wound signaling into the human timescale’.
We are happy to announce the forthcoming workshop entitled “Roadmap for EvoDevoMec”, Nov. 2nd – 5th, 2026, Université Paris Cité.
The link with evolutionary history of an organism is key to understand embryonic development. Much of the focus has been so far on genetic circuitry. However, it has become increasingly clear that physical constraints are an essential aspect at the basis of morphogenetic processes. We have now reached an exciting stage where it becomes possible to integrate mechanical considerations in our view of how development has been shaped during evolution: EvoDevMec.
This new approach has recently raised great interest, but biological questions still need to be reformulated with precision. In addition, due to its integrative nature, it poses a scientific and technical challenge: What type of experimental systems could be used? How could we define experimental proof of concepts? How can theoretical biophysical models help integrate various questions and approaches?
We aim at discussing these questions in an informal context, i.e. with chalk talks. The workshop is limited to 30 persons, from PhD students to senior scientists from various countries. We will welcome biologists using a breadth of models (from unicellular organisms to animal and plant models), physicists (theoreticians, numericians, experimentalists), and mathematicians.
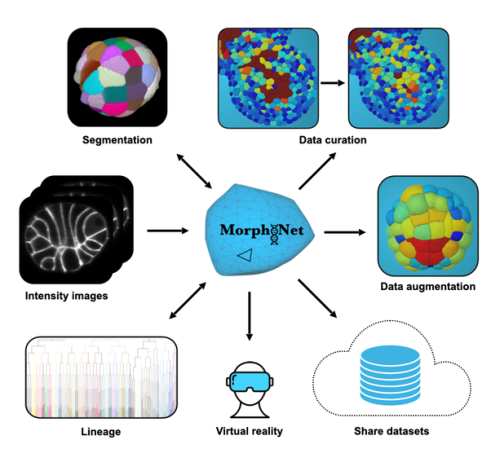
Our ‘Featured resource’ series aims to shine a light on the resources that support our research – the unsung heroes of the science world. In this post, we learn about the data and functionalities available at MorphoNet 2.0, and hear about new initiatives they are developing.
MorphoNet 2.0: breaking the glass ceiling of 3D+time bioimage curation
Modern microscopy now allows us to image living systems in three dimensions over time. From early embryonic divisions to complex tissue morphogenesis, we can follow every cell with remarkable spatial and temporal resolution.
Yet a critical bottleneck remains. Even with powerful AI-based segmentation tools, such as Cellpose or StarDist, errors are inevitable. In large 3D+time (3D+t) datasets, a seemingly small error rate quickly translates into thousands of mis-segmented cells, broken lineages, and ultimately misleading biological conclusions.
Correcting these errors — a process known as curation — is often so time-consuming that it becomes the real “glass ceiling” of quantitative developmental biology.
MorphoNet 2.0 was designed to break this glass ceiling.
From segmentation to curation
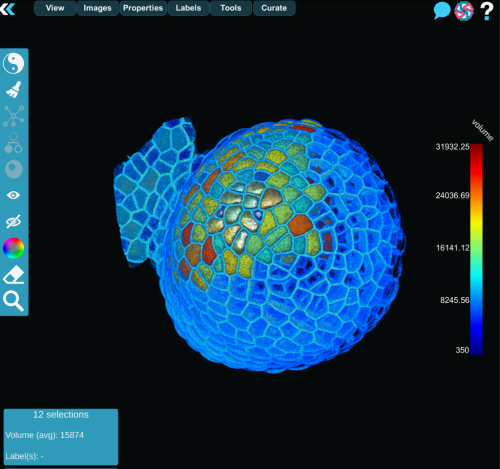
MorphoNet 2.0 is a major evolution of the original platform. Rather than proposing yet another segmentation algorithm, it focuses on a problem that is just as critical, but far less addressed: how to efficiently assess, correct, and validate 3D and 4D segmentations at scale.
The key idea is simple: automated segmentation is only useful if biologists can trust and refine its output. MorphoNet 2.0 makes this process fast, efficient, interactive, and accessible to non-programmers.
A hybrid architecture built for 3D+t data
Unlike the original web-based MorphoNet, version 2.0 is a standalone application designed to run on standard research workstations and laptops. Its architecture deliberately bridges two complementary worlds:
a high-performance 3D interface, powered by the Unity game engine, for smooth exploration of thousands of objects in real time
a Python backend integrated with powerful scientific image-processing libraries, enabling advanced image analysis, editing, and AI-based segmentation directly on raw data.
Letting the data highlight its own problems
A major challenge in large 3D datasets is simply knowing where to look. Manually inspecting every cell is unrealistic.
MorphoNet 2.0 addresses this by computing unsupervised quality metrics for each segmented object, such as volume, shape regularity, boundary intensity, or temporal stability. These properties are projected, as color maps, directly onto the 3D meshes or cell lineages, turning potential errors into visible outliers.
While these metrics are not absolute measures of correctness, they act as intelligent guides, directing experts toward regions that deserve closer inspection.
Curation as a local, interactive process
Once a potential error is identified, MorphoNet 2.0 enables rapid local, targeted corrections rather than global reprocessing.
This is achieved through a modular plug-in system that allows users to perform a wide range of operations, including (but not limited to):
re-segment specific regions using AI or classical methods
split fused cells or merge over-segmented fragments
remove specifically small objects
propagate corrections across time in 3D+t datasets
edit and repair cell lineages.
Because plug-ins operate only on user-defined regions of interest, most corrections take seconds rather than hours. This transforms curation from a batch process into a true human-in-the-loop workflow, with immediate visual feedback.
MorphoNet 2.0 provides an open plug-in architecture that enables contributors to develop and share custom plug-ins tailored to their own segmentation, tracking, or lineage-repair challenges.
Tested on real biological datasets
This paper demonstrates MorphoNet 2.0’s successful use on five previously published datasets spanning insects, plants, nematodes, echinoderms, and ascidians. These case studies show how the platform can:
reveal hidden segmentation errors in datasets long considered “finished”
significantly improve segmentation quality through iterative correction
polish cell lineages to a level suitable for studying subtle biological variability.
In one example, rare lineage errors scattered across thousands of cells were identified and corrected in a few hours — even in datasets comprising several thousand segmented objects — a task that would have been practically infeasible with traditional tools.
Why this matters for AI and data reuse
High-quality 3D ground truth data are essential for training and benchmarking modern AI models. Yet producing such datasets is extremely costly when curation tools do not scale.
By making 3D+t curation feasible, traceable, and accessible to biologists, MorphoNet 2.0 directly addresses this gap. It helps turn raw automated outputs (“silver truth”) into reliable, reusable datasets (“gold truth”) that can support reproducible analysis, fair algorithm comparison, and community challenges.
Who is MorphoNet 2.0 for?
MorphoNet 2.0 is designed for:
developmental and cell biologists working with complex 3D or 4D imaging data
imaging facilities producing reference datasets
researchers developing or benchmarking segmentation and tracking algorithms.
No programming skills are required to use the platform, but its open, Python-based plug-in architecture allows advanced users to extend it and share new tools with the community.
Looking ahead
MorphoNet 2.0 positions complex 3D curation as a fast and manageable activity rather than a never-ending technical burden. By combining intuitive 3D interaction, unsupervised quality assessment, and local correction, it offers a practical solution to one of the most persistent bottlenecks in modern bioimage analysis.
I recently attended the BSDB Spring Meeting, and decided to spend most of my time at the conference sketching. The result is this illustrated summary – featuring portraits of almost all the speakers alongside drawings of elements from their talks, whether that’s a model organism, a signalling pathway, or a particularly striking image from their work.
I initially felt quite anxious and self-conscious to be drawing in public. After all, most of the time when I’m creating art at home, it tends to look rough right up until the last moment when it all comes together. I also normally paint from static references, which conference speakers definitely are not! But, I finally decided to take the leap after numerous conversations with veteran conference illustrator Alex Cagan, who urged me to go for it. Once I started, I could feel that each drawing was turning out a little better than the last. I have always been a visual learner and definitely felt more engaged while sketching, rather than frantically scribbling notes as I would normally do!
What started as 30+ separate illustrations on my iPad slowly turned into this after I got home from the conference. My original intention was to share each as a separate piece. However, none of the illustrations felt complete enough for this (I will definitely have to work on my drawing speed in the future), so my solution was to combine them all into one large piece.
This isn’t intended as a scientific summary, more as a memento or a snapshot of what it felt like to sit in those sessions, surrounded by so much brilliant developmental biology. If you spot yourself in there, I hope I have done you justice! And I apologise if I missed out on drawing you – at times, I couldn’t quite keep up.
I’m interested in using illustration to share the joy of developmental biology, as I’ve tried to do here, but I also think it can be a wonderful tool for communicating complex scientific ideas to those who might not otherwise engage with them. This conference reminded me how much exciting work there is to communicate. Thanks to all the speakers and organisers for making it such an enjoyable meeting!
This year the popular Placental Biology Course returns online from 14 to 18 September.
This online course is designed for a diverse audience, including students, postdoctoral researchers, established academics, medical and veterinary healthcare professionals, and industry specialists with an interest in the latest developments in placental biology.
The programme includes pre-recorded lectures and practical sessions delivered by leading experts in the field, allowing participants to engage with cutting-edge research at their own pace. Each day also includes live Q&A sessions, offering a valuable opportunity to interact directly with speakers and deepen understanding.
In addition, attendees can take part in Fellowship Workshops and informal Meet and Greet sessions to build connections within the community. Participants are also invited to submit abstracts for consideration in flash talks or virtual poster presentations, providing a platform to showcase their own work.
Join us in April to learn about the three papers named as finalists of Development’s 2025 Outstanding Paper Prize. In this webinar two of the papers will be presented by its first author and chaired by Deputy Editor, Steve Wilson. Learn more about the finalists in our Editorial and read the full shortlist of nominated papers in Development’s subject collection.
Wednesday 29 April – 15:00 BST (UTC+1)
Xi Yang (Purdue University, USA) ‘Reprogramming single cells into multicellular meristems: insights into sex‑type conversion and de novo meristem formation in the fern Ceratopteris’
Bénédicte Lefèvre (Institut Curie, France) ‘Evolution of a novel left-right asymmetry in organ size by co-option of a tissue rotation process’
The third finalist, Christopher De Bono (INSERM, France),will present ‘Multi-modal refinement of the human heart atlas during the first gestational trimester’ later in the year.
At the speakers’ discretion, the webinar will be recorded to view on demand. To see the other webinars scheduled in our series, and to catch up on previous talks, please visit: thenode.biologists.com/devpres
Our February webinar featured three early-career researchers studying neural development. Here, we share the talks from Joaquín Navajas Acedo (University of Basel) and Carlo Donato Caiaffa (Universidade de São Paulo).
I have long been interested in understanding how development emerges at the intersection of molecular and spatial organization. On one hand, decades of work have identified the genes and signaling pathways that control cell fate. On the other hand, classical embryology and biophysics have revealed how cells move, change shape, and assemble into tissues. Yet, directly connecting these two layers—linking gene expression to spatial patterns and morphogenesis at the scale of a whole embryo—has remained challenging.
When I joined the Schier lab in March 2020, single-cell RNA-seq approaches had already enabled the reconstruction of developmental trajectories with remarkable molecular detail [1]. For the first time, we could computationally line up cells along developmental paths and begin to understand how cell fates emerge at the whole-embryo level. But something always felt missing to me: these trajectories existed in abstract space, detached from the physical embryo. We could describe where cells were going, but not where they actually were.
At the same time, there was growing interest in spatial transcriptomics within the lab and through our membership in the Allen Discovery Center for Cell Lineage Tracing. This created an opportunity to bridge molecular and spatial information in developing systems. With my background in imaging and technology development, I was particularly drawn to the idea of building a method that could map gene expression across whole embryos, while preserving spatial organization at high resolution.
Technically, the initial setup went relatively smoothly. Ahilya Sawh (then in Susan Mango’s lab at the Biozentrum, now leading her own group at the University of Toronto, Canada) had previously established a FISH-based system for chromosome tracing in C. elegans [2, 3]. With support from the Biozentrum Imaging Core Facility, we adapted this system onto a Nikon microscope, making it more accessible for biological applications. Having this foundation in place was reassuring—it meant that the challenge ahead was not starting from zero, but rather pushing something promising to its limits.
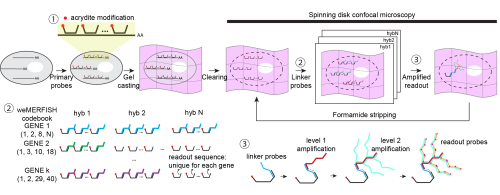
Fig.1 weMERFISH imaging platform for whole-embryo spatial transcriptomics. The weMERFISH system integrates a spinning disk confocal microscope with a microfluidic setup to enable automated, multiplexed imaging over extended periods.
Making it work in a whole embryo
Early on, we realized that if we wanted to understand gene expression in a whole embryo, measuring just a handful of genes would not be enough. We needed to look at hundreds of genes at the same time, but without an impractical number of imaging rounds. This led us to explore MERFISH, which uses a combinatorial barcode system to identify genes across multiple rounds of imaging and allows many genes to be read out efficiently [4, 5]. In practice, it felt like a good balance between scale and feasibility for what we wanted to do.
While the foundation was in place, adapting MERFISH to whole embryos still required several key innovations. An important part of this effort was our close collaboration with Bogdan Bintu at UCSD in the USA. During his time in Xiaowei Zhuang’s lab at Harvard University, Bogdan developed high-throughput imaging approaches that combine MERFISH with 3D chromatin organization [6], contributing deep experience in both experimental and computational aspects. He had already begun implementing important technical improvements and generously shared his strategies with us, as well as providing essential support for the instrumentation and computational pipeline.
In the end, making this work required combining several key improvements, each addressing a different limitation of the system:
• Stability during long-term imaging
Whole-embryo MERFISH imaging can take weeks, as the sample must be imaged plane by plane, region by region, and across multiple hybridization rounds. Early on, this long-term imaging became one of the most frustrating bottlenecks. Using conventional approaches, in which mRNAs are anchored to a gel and repeatedly probed, we saw signals gradually fading over time. Even under carefully controlled RNase-free conditions, degradation was unavoidable.
The solution came from rethinking the problem: instead of anchoring RNA, we anchored the primary DNA probes using acrydite modifications. This seemingly simple shift made a huge difference. The signal remained stable even after a month of continuous imaging, and we could use harsh stripping conditions (80–100% formamide) without worrying about losing the probes. It was one of those moments where a technical fix suddenly makes everything feel possible.
• Seeing deeper without losing signal
Imaging deep tissues introduced another challenge. Spinning disk confocal microscopy is essential for thick samples, but it comes with reduced signal compared to widefield imaging. We initially explored computational approaches to recover signal, but in practice, signal and noise often looked too similar to confidently separate.
Instead, we turned to a physical solution: branching amplification. By boosting the signal at the molecular level, we were able to image more reliably, reduce exposure time, and make the data much more interpretable, especially in deeper regions of the embryo.
• Designing for flexibility
Another challenge was making the method usable in practice. Traditional MERFISH requires encoding the combinatorial barcode directly into the primary probes, effectively locking the experiment into a fixed readout strategy.
We wanted something more flexible. By introducing a gene-specific linker system, we decoupled probe design from readout. This means that researchers can design large probe libraries first, and decide later how to read them out—sequentially for a few genes, or combinatorially for many. This flexibility turned out to be important not just technically, but psychologically: it lowers the barrier to trying the method in the first place.
Fig. 2 Workflow and design principles of weMERFISH. Primary probes with acrydite modifications hybridize to mRNAs and are anchored in a gel, enabling stable imaging over many cycles. Gene-specific linker probes and sequential fluorescent readout identify transcripts through a combinatorial barcode. Signal is enhanced by branching amplification for imaging in whole embryos.
When the data started to speak
Once we had the data, the analysis moved quickly. I remember feeling a mix of excitement and disbelief: after spending three years building the system, suddenly everything was there at once. The work was tremendously accelerated by the incredible team behind it: Jakob El Kholtei and I co-developed the weMERFISH method and the data processing pipeline. KJ Jenie, an exceptionally talented undergraduate at the time in Bogdan’s lab, built the MERFISHEYES website (https://schier.merfisheyes.com), making the dataset accessible and explorable. A key component of the project came from my postdoctoral colleague Jialin Liu, who had generated a comprehensive scMultiomics dataset of zebrafish development to study the regulatory logic of cell type specification [7], and very generously made it available to us. Integrating his scMultiomics data with the weMERFISH data allowed us to comprehensively map gene expression and chromatin accessibility in space, creating a multiomic atlas. Mariona Colomer-Rosell performed the analysis of these multiomic data and helped uncover principles of tissue-specific gene regulation at the whole-embryo level. Bringing together these different pieces was essential for turning the dataset into something we could truly interpret.
One of the most fascinating aspects for me was the concept of “time” in development. In single-cell data, we often reconstruct “pseudotime” trajectories, but seeing these trajectories mapped into real space was incredibly satisfying. Along the zebrafish tail, for example, we could directly observe the progression from progenitors to differentiated cells as a spatial gradient. It was one of the first moments where the abstract and the physical truly aligned.
We also applied a spatial version of RNA velocity [8, 9], using nuclear versus cytoplasmic transcripts as a proxy for transcriptional dynamics. What surprised us was that, especially in early development, the inferred transcriptional dynamics mirrored physical cell movements during morphogenesis. At first glance, this feels intuitive. But the underlying mechanisms are very different: transcriptional regulation and cell movement are controlled by distinct processes. The fact that they align so closely suggests a deeper coupling between gene expression dynamics and morphogenesis. This was one of those observations that stayed with me, because it hints at something fundamental that we don’t yet fully understand.
Another memorable part of the journey was the path from preprint to publication. When we first posted the work on bioRxiv and launched the MERFISHEYES website, the response was immediate and very encouraging. People started exploring the data, reaching out with questions, and even visiting the lab to learn how to set up the method. Seeing the dataset being used so quickly made us realize that it could become a resource for the community much earlier than we had expected.
At the same time, the peer review process pushed the work in important ways. The reviewers appreciated the technology and the dataset, but also challenged us to go further, especially to better connect the method to biological questions and to take fuller advantage of the multimodal data. Addressing these comments led to substantial additions and improvements throughout the paper. We expanded the analysis of subcellular transcript localization, strengthened the RNA velocity framework, benchmarked data integration methods more rigorously, and added new analyses such as cell–cell communication.
Perhaps most importantly, the revision motivated us to develop MERFISH-FATE in collaboration with Guoqiang Yu’s group (Tsinghua University, China), integrating spatial transcriptomics with live imaging to directly link gene expression changes to morphogenetic movements. Specifically, we mapped corresponding regions between a weMERFISH embryo and a live-imaged embryo at early gastrulation, where cells had been tracked throughout development. We then followed these trajectories forward and mapped the descendant cells back to their corresponding regions at mid-gastrulation, effectively connecting gene expression patterns across time.
This became a great extension of the story and shifted the paper from a largely static atlas into a more dynamic view of development. We then spent months simply looking at how patterns evolve—scrolling through images, comparing stages, trying to build intuition. Across many genes, we saw surprisingly complex dynamics. One example is tbxta, which is expressed at the embryonic margin at both early and mid-gastrulation. It would be natural to assume this reflects simple inheritance. But when we incorporated cell dynamics, we found that some cells activate tbxta while others turn it off. What looked like a static pattern was actually the result of dynamic and opposing processes. Moments like this made me appreciate how much information is lost when we only look at snapshots, and how powerful it is to connect gene expression with cell behavior. This is a direction we are now continuing to explore in more depth in a recent preprint describing fate mapping in zebrafish embryogenesis and beyond [10].
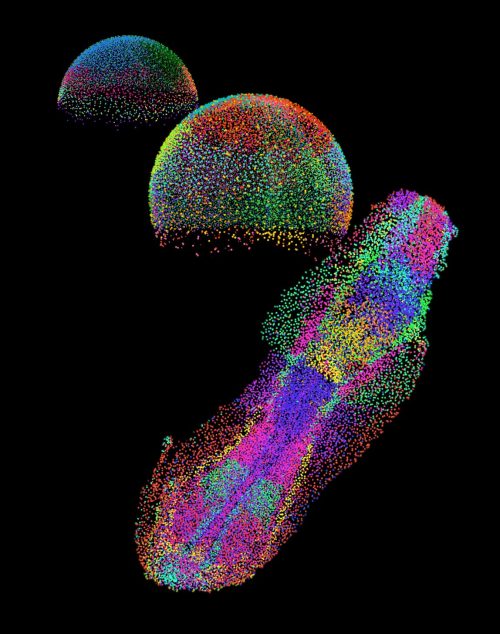
Fig. 3 An embryo in the making. Each color shows a distinct group of cells as they organize during early development of a zebrafish embryo.
Looking ahead: from description to understanding
weMERFISH provides a way to map gene expression and cellular states across intact, developing tissues in 3D. For me, a particularly exciting direction is to extend this approach to larger and more complex systems: whole organs, organoids, and beyond. Moving into these systems will allow us to study how spatial gene expression patterns scale with size and geometry, and how these patterns are adapted across evolution.
At the same time, a major direction in the field, in my view, is the integration of multiple modalities. In this work, we combined weMERFISH with chromatin accessibility and embryo morphogenesis, and this naturally raises broader questions: how does gene expression relate to 3D genome organization? To protein distribution? To lineage history and cell behavior? Each of these layers captures a different aspect of cellular identity, and I believe only by combining them can we begin to understand how cells make decisions in their native context.
This is also a direction I am eager to pursue in my own future work. These multimodal datasets are not just richer—they fundamentally change the type of questions we can ask. Instead of only describing patterns, we can begin to build models that explain how these patterns arise. We can start to ask causal questions: which molecular features predict a cell’s future behavior? Which spatial contexts bias fate decisions? And how conserved are these relationships across tissues and embryos?
Ultimately, I am particularly interested in understanding how variability between individual cells gives rise to robust and reproducible tissue structures. Development is remarkably reliable, despite underlying variability. Technologies like weMERFISH bring us closer to uncovering these principles, and to understanding how a single fertilized egg reliably gives rise to a complex organism.
Access the article:
Wan, Y., J. El Kholtei, I. Jenie, M. Colomer-Rosell, J. Liu, Q. Zhang, J. Navajas Acedo, L. Y. Du, M. Codina-Tobias, M. Wang, W. Zheng, E. Lin, T. H. Chuang, O. Mayseless, A. Sawh, S. E. Mango, G. Yu, B. Bintu, and A. F. Schier. 2026. “Whole-embryo spatial transcriptomics at subcellular resolution from gastrulation to organogenesis.” Science 391 (6790): eadt3439. https://doi.org/10.1126/science.adt3439.
References
Farrell, J.A., et al., Single-cell reconstruction of developmental trajectories during zebrafish embryogenesis. Science, 2018. 360(6392).
Sawh, A.N., et al., Lamina-Dependent Stretching and Unconventional Chromosome Compartments in Early C. elegans Embryos. Mol Cell, 2020. 78(1): p. 96–111 e6.
Sawh, A.N. and S.E. Mango, Multiplexed Sequential DNA FISH in Caenorhabditis elegans Embryos. STAR Protoc, 2020. 1(3): p. 100107.
Chen, K.H., et al., RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science, 2015. 348(6233): p. aaa6090.
Moffitt, J.R. and X. Zhuang, RNA Imaging with Multiplexed Error-Robust Fluorescence In Situ Hybridization (MERFISH). Methods Enzymol, 2016. 572: p. 1–49.
Su, J.H., et al., Genome-Scale Imaging of the 3D Organization and Transcriptional Activity of Chromatin. Cell, 2020. 182(6): p. 1641–1659 e26.
Liu, J., et al., Decoding the regulatory logic of specification and differentiation during vertebrate embryogenesis. bioRxiv, 2024.
La Manno, G., et al., RNA velocity of single cells. Nature, 2018. 560(7719): p. 494–498.
Bergen, V., et al., Generalizing RNA velocity to transient cell states through dynamical modeling. Nat Biotechnol, 2020. 38(12): p. 1408–1414.
Wang, M., et al., High-Fidelity Long-term Whole-embryo Lineage and Fate Reconstruction by Iterative Tracking with Error Correction. bioRxiv, 2026: p. 2026.03. 12.711203.
The laboratory of Assistant Professor Dr. Jette Lengefeld is inviting applications for a position as Laboratory Technician who is extensively experienced in experimental mouse work.
About the group The Lengefeld laboratory is working on understanding how the failure to regenerate tissue with age is a major health issue. A contributor to this decline is the loss of stem cell function. Despite the essential role of stem cells, it is still unclear how they fail to maintain their functions during aging and disease. We discovered a new aspect of stem cell aging in vivo: cellular enlargement. With age and damage, stem cells increase in size causing their functional decline. However, we are only beginning to understand how size impacts stem cell fitness and the physiological importance of this process remains unsolved.
The position This position includes several responsibilities. The main activities are listed below:
1. Supporting research projects of lab members, with a strong drive to intellectually contribute. Tasks will include mouse genotyping, mouse blood sampling, mouse monitoring and communication with mouse facility staff. We expect you to fully understand the experimental design and underlying reasons for performing the experiment.
2. Performing administrative tasks for group leader, research meetings and bills.
3. Active intellectual contribution to research discussions such as lab meetings, journal clubs, and project discussions.
Required qualifications and experience – Extensive experience in experimental mouse work and authorization to work with mice is required (please do not apply if not applicable).
– Minimum required education: Master’s degree.
– Experience in hematology will be prioritized.
– The candidate must be fluent in English and Finnish language skills are an advantage.
– The candidate is collaborative, communicative, and comfortable working in an international and multidisciplinary environment.
Salary and contract The position is offered for a fixed term of one year starting in May 2026 or as negotiated. Salary will depend upon the applicant’s level of skills, knowledge, and abilities and is based on the university salary system. A trial period of 6 months will be applied.
Interested? Application should include the following documents: cover letter (max 1 page), CV of max. 4 pages, full publication list, and names of three references.
The deadline for the applications is 30.04.2026, but the positions will be filled immediately once suitable candidates have been identified.
Please submit your application, together with the required attachments, through the University of Helsinki electronic recruitment system by clicking the “Apply for the position” – link below. Internal applicants (i.e., current employees of the University of Helsinki) must submit their applications by logging in to the SAP system: https://msap.helsinki.fi. For technical support regarding the recruitment system, please contact rekrytointi@helsinki.fi.
If you have any questions about the position, please do not hesitate to contact postdoc Dr. Emilie Cerezo emilie.cerezo(at)helsinki.fi.
Link to apply: https://jobs.helsinki.fi/job/Helsinki-Laboratory-Technician%2C-group-Lengefeld/1356106757/
 (No Ratings Yet)
(No Ratings Yet)



 (6 votes)
(6 votes)