Applications are open for the Wellcome Trust funded four year PhD programme in Developmental Mechanisms at the University of Cambridge. We are looking for talented, motivated graduates or final year undergraduates, and are keen to attract outstanding applicants in the biological sciences, who are committed to doing a PhD. We are able to fund both EU and non-EU students.
Closing date: 4th January 2018
For more details about the application process and the programme please see the website:
A fundamental aspect of vertebrates is their external bilateral symmetry, which has to some extent shaped evolutionary success. Not only is beauty associated with symmetry, enhancing an individual’s chance of mating but also, symmetry in the legs will help an animal flee from a hunter (Enquist and Arak, 1994; Johnstone, 1994; Holló and Novak, 2012). However, a remarkable feature of the vertebrate body beyond this external symmetry is the asymmetric disposition and morphology of the internal organs relative to the left-right (L-R) axis. These asymmetries are essential for optimal organ packaging and function, yet it has long remained a mystery as to how this asymmetry is achieved. The initial symmetry is first broken in the vertebrate embryo at the level of the L/R organiser during early gastrulation. As a result, asymmetric information is transmitted to the lateral plate mesoderm (LPM), restricting the expression of Nodal and its downstream target Pitx2 to the left side of the embryo. The Nodal-Pitx2 pathway, which is conserved in all vertebrates, is instrumental in controlling L/R asymmetry (Raya and Izpisua Belmonte, 2006). This left-handed information is repressed on the right-hand side by the inducer of the epithelial-mesenchymal transition (EMT), Snail1. It has remained unclear whether an equivalent right-handed pathway provides instructive information to the right LPM. Laterality defects are associated with some important diseases in humans. Thus, the mechanisms underlying the establishment of L/R asymmetry are clearly of interest to developmental biologists and fully understanding these events will have significant biomedical implications.
New Findings
The mechanisms that regulate the EMT in development and disease have for long been of particular interest to our lab. When screening for genes expressed in the LPM of chick embryos, we found the Prrx1 transcription factor to be a particularly potent inducer of the EMT, and like Snail1, not only in embryos but also in cancer cells (Ocaña et al., 2012). When the expression of these genes was studied in more detail, we realised that Prrx1 is transiently expressed asymmetrically in the same temporal window as Snail1. Moreover, and as we also described previously for Snail1 (Morales et al., 2007), the expression of Prrx1 was stronger on the right than on the left side of the embryo. Likewise, prrx1a transiently appears to be asymmetrically distributed in zebrafish embryos during a similar developmental window as in the chick embryo, again with higher levels on the right-hand side. Therefore, with great expectation, we tested whether the asymmetric distribution of Prrx1 influenced organ laterality, focusing on heart positioning as this is the first clear indication of morphological L/R asymmetry in the embryo. The fastest and easiest way to tackle this question was to knock-down prrx1a expression in the zebrafish and we found that at 48 hpf, the majority of the morphant embryos developed mesocardia and the dextral looping typically observed in embryos was completely abrogated. To determine whether this effect was conserved in chicken, we performed loss of function experiments by electroporating RNAi against Prrx1 bilaterally into cardiomyocyte precursors. Prrx1 downregulation provoked a similar effect, with heart mesocardia being the main phenotype observed. Thus, it seemed that in both the fish and chicken asymmetric Prrx1 expression was required for heart laterality.
Figure 1 Prrx1 L/R asymmetric expression in zebrafish and chick embryos. Left panels, ventral view of a maximum intensity projection of whole zebrafish embryos at 30 hpf (upper) and z-plane section of confocal images immunostained to visualize Prrx1 protein expression. Note the higher number of Prrx1+ cells in the pericardium on the right hand side. Right panels, ventral view of a 13 somite-stage whole mounted chick embryo and transverse section showing the asymmetric L/R Prrx1 protein expression, higher on the right sinus horn (RSH). Nuclei were stained with DAPI (blue). Scale bars, 100 μm. AIP, Anterior Intestinal Portal; E, Eyes; HT, Heart Tube; L or R SH, Left or Right Sinus Venosus Horns; P, pericardium.
Given the conserved asymmetric expression of Prrx1, stronger on the right-hand side, and its role during heart looping, we reasoned that an instructive pathway may convey information to the right-hand side, in addition to the left pathway. Support for the existence of a right-handed program came from studies into mild Snail1 downregulation on the right-hand side, which provoked heart looping defects in the chick embryo without affecting Pitx2 expression (Patel et al., 1999). In addition, the development and position of the proepicardium, a transient specifically right-sided structure in frogs and avians, was affected by altered Snail1 expression but it was not after aberrant bilateral Pitx2 expression (Schlueter and Brand, 2009). Furthermore, zebrafish and mice carrying mutations in Pitx2 do not display heart looping defects (Campione et al., 2001; Ji et al., 2016). Together, these data suggest that heart laterality may be driven by a dominant right-handed pathway. As such, we speculated that reversing the levels of Prrx1 expression, so that higher levels were expressed on the left rather than the right-hand side, would lead to the development of embryos with reverse looping.
To test our hypothesis, we took advantage of the electroporation technique that allows unilateral manipulation of gene expression in chick. We studied the effect of Prrx1 gain-of-function in the left LPM and we found that a significant proportion of the embryos displayed reverse heart situs. These data indicate that the transiently stronger expression of Prrx1 in the right LPM was sufficient to drive heart looping. To gain further insight into the mechanism underlying this transient asymmetric expression of Prrx1 and how it affected heart looping, we started to precisely characterise the cell populations that contribute to heart development and that express this gene. This involved developing an antibody against Prrx1 and performing dual or multiple immunolabelling of cell populations associated with the heart using different markers, carrying out an exhaustive confocal analysis. As a result, we found that Prrx1 was not expressed in the primary heart tube (PHT) but that it was in fact expressed by a population of cells lateral and posterior to the cardiac venous pole.
The finding that another EMT inducer, in addition to Snail1, was strongly expressed in the right LPM, also influencing heart situs, suggestsed to us that some common features could exist between L-R pathways and the EMT program. As such, we were prompted to study whether differential L-R cell movements promoted by Prrx1 may drive heart looping. To investigate the movement and fate of the cells that express Prrx1 and that contribute to the posterior pole, new tools had to be generated and thus, we first addressed this question in the zebrafish. We generated a tbx5a-reporter transgenic line Tg(tbx5a:eGFP) as the cells expressing this gene contribute to both the PHT and the posterior pole of the heart (Ahn et al., 2002). In this tbx5a-reporter line there was complete co-localization between eGFP and Prrx1 in a subpopulation of the LPM cells, in the region of the cardiac precursors, which validated the use of these animals. When cell movements were followed in this reporter line, time lapse recordings of embryos from 28 to 48 hpf (during which time heart looping occurs) indicated an asymmetric migration of the Tbx5/Prrx1 double positive cells at the posterior pole of the heart. Significantly, there was a higher contribution of cells from the right than from the left-hand side, which correlates perfectly with the asymmetric expression of Prrx1 driving a different left/right cell contribution. In fact, prrx1a downregulation impaired this asymmetric cell migration of posterior pole cells and leads to mesocardia.
These observations raise the question as to whether these cells contribute to heart positioning and morphogenesis. Lineage tracing in zebrafish embryos demonstrated that Prrx1 expressing cells contribute to the PHT at the time of the heart looping, yet this Prrx1 expression is downregulated concomitant with their incorporation into the heart tube. Interestingly, the size of the atrium was reduced in the Prrx1a morphants, indicating that Prrx1 expression by cardiac progenitors is also required for heart morphogenesis.
The heart looping defects observed when Prrx1 was downregulated were compatible with studies describing the asymmetric contribution of cells from the right and left of the embryo to the heart after the formation of the PHT (Taber et al., 2010; Dominguez et al., 2012). Thus, we thought that differential left-right cell movements driven by Prrx1 could generate asymmetric forces and tension that would be stronger from the right. Such forces might eventually lead to the initial leftward bending of the posterior pole of the heart and the subsequent dextral torsion. In accordance with this hypothesis, cells in the tbx5a reporter could be seen migrating from the right-hand side to the posterior pole at the heart looping stage, apparently forming a structure reminiscent of a cable. Since forces in developing tissues are usually controlled by actomyosin bundles, we visualized actin stress fibres in vivo and while cardiac looping was normally accompanied by the formation of an actomyosin cable directed towards the posterior pole, this was not the case in the Prrx1a morphant embryos. Moreover, laser ablation of Prrx1/Tbx5 expressing cells on the right but not on the left side of the embryo prevented heart lateralization. These experiments demonstrate that asymmetric tension, more intense on the right-hand side, drives heart looping. Interestingly, we found that this mechanism was conserved in the chick embryo. Collectively, these data indicate that as in the fish, leftward displacement of the posterior pole and the subsequent dextral looping is driven by an actomyosin-dependent mechanism.
Having found that the asymmetric expression of Prrx1 plays a key role in heart lateralization, both in zebrafish and chick embryos, the next issue was to place this transcription factor it in the signalling pathways already known to be involved in L/R asymmetry. We knew that BMP could induce Prrx1 expression in the chicken embryo LPM (Ocaña et al., 2012), where it also induces Snail1 to repress Pitx2 expression (Raya and Izpisua Belmonte, 2006). In loss- and gain-of-function experiments performed on zebrafish and chick embryos, we confirmed that Prrx1 was activated by BMP and repressed by Nodal signalling. Hence, heart looping is driven by a BMP-mediated pathway that promotes strong Prrx1 expression, and that is repressed on the left by Nodal.
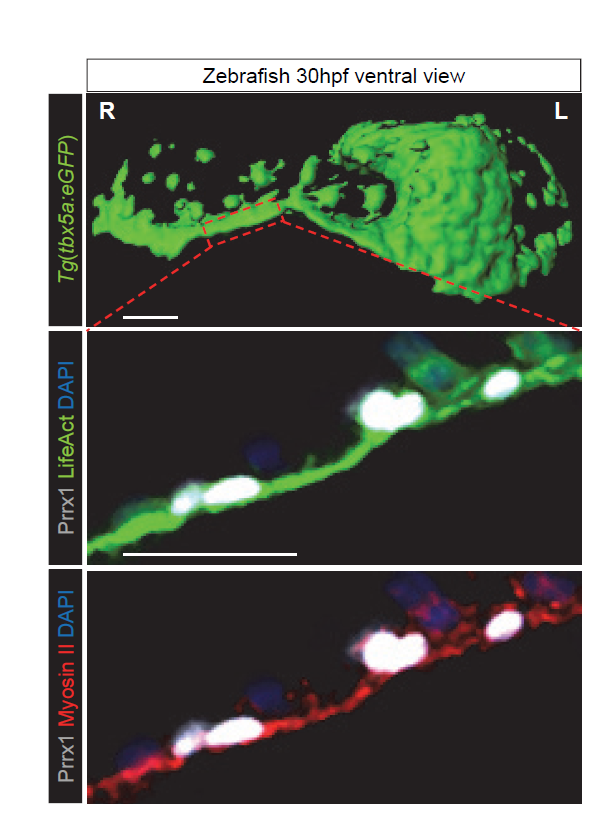
Figure 2 Prrx1 asymmetric expression generates actomyosin-mediated differential forces. Snapshot of surface rendering image from a Tg(tbx5a:eGFP) embryo at 30 hpf showing the presence of a “cable-like” structure on the right-hand side during heart looping stage (upper panel). High power confocal images of cells located in positions similar to the area boxed in a Tg(actb2:myl12.1-mCherry) reporter zebrafish embryo to visualise myosin II (red). The embryo was injected at 1 cell-stage with LifeAct:GFP mRNA to visualise F-actin (green) and subjected to immunohistochemistry for Prrx1 expression (white). Cell nuclei were stained with DAPI (blue). Scale bars, 50 μm.
A surprising evo-devo twist
Having shown the implication of Prrx1 in heart looping in both the fish and chick embryo, we turned our attention to the mouse embryo. At first, it was disappointing that our model did not seem to fit well, as there was no heart looping defect evident in Prrx1 mutant mice (Bergwerff et al., 2000). However, we knew that Snail1 mutants develop heart looping defects (Murray and Gridley, 2006) and thus, we compared Prrx1 and Snail1 expression side-by-side in mouse and chick embryos. In the territories relevant to heart looping, we found an interchange of expression patterns between these two EMT inducers (Prrx1 and Snail1). While chick embryos displayed asymmetric Prrx1 expression in the ventral posterior pole of the heart, mice displayed asymmetric Snail1 expression along with an accumulation of F-actin fibres in this territory, but no Prrx1 expression. As such, these results offered a convincing explanation for the heart laterality defects observed in Snail1 mutant mice and the lack of such defects in the Prrx1 mutants. Thus, in terms of heart laterality, it is Snail1 in the mouse that seems to carry out the role of Prrx1 in the fish and chick. While this observation may appear surprising, we had previously observed other interchanges in the lab, such as those between Snail1 and Snail2 expression at other sites in the chick and mouse embryos (Locascio et al., 2002).
General conclusions
We have identified a prominent right-handed pathway that is driven by BMP signalling and that is in turn repressed on the left-hand side by Nodal. This right-hand signalling induces asymmetric L/R activation of different EMT transcription factors, Prrx1 in fish and chick, and Snail1 in the mouse embryo, which provoke asymmetric cell movements and forces that are stronger on the right-hand side of the embryo. In fact, these asymmetric forces induce the leftward displacement of the posterior pole and dextral looping of the heart in an actomyosin-dependent manner. We are very excited as to how these studies have progressed, as they have allowed us to unravel a basic mechanism that has been conserved in vertebrates and that controls heart looping. Furthermore, this mechanism could help us better understand the congenital heart diseases that are related to heart laterality in humans.
The goal of this project is to engineer therapeutically active islet-like aggregates for future cell therapy phase 1 trials in Type 1 Diabetes (T1D)
Job description
The laboratory technicians will be responsible for the maintenance of the human pluripotent stem cell (hPSC) lab. The work will include expansion of human embryonic and induced pluripotent stem cells into characterized (e.g. marker analysis and karyotyping) hPSC line batches, development of new methods and protocols for hPSC maintenance and differentiation, transfections of hPSCs and basic characterization of hPSCs (undifferentiated and differentiated) by immunohistochemistry, qPCR and FACS. The laboratory technicians will secure the quality and reproducibility of the hPSCs to be used by scientists in the group. The candidates will work together with a dedicated team of scientists who together will tackle bottle-necks towards implementing the first phase 1 clinical trial in T1D. The working hours are 37 hours per week. The positions are time limited to the end of 2020 with a possibility of extension.
Qualifications
Highly motivated and ambitious candidates are encouraged to apply. The positions require solid experience with cell culture, including transfection (traditional and virus-based methods) and gene expression is necessary. Experience in hPSC culture, molecular biology and immunohistochemistry is required. Knowledge of cell biology, developmental biology and morphology is an advantage. The work is independent and demands flexibility and accuracy. Further, you must have good interpersonal skills and good command of English.
Terms of salary, work, and employment
The employments are planned to start as soon as possible upon agreement with the chosen candidate. The place of work is at DanStem, University of Copenhagen, Blegdamsvej 3B, Copenhagen. The positions are time limited to the end of 2020 with a possibility of extension.
Terms of appointment and salary are in accordance with the agreement between the Danish Government and HK-STAT (Danish Technician Association). The position will be at the level of salary group 5 with the possibility to negotiate due to qualifications and experiences.
Application
An application for any of the positions should be submitted electronically by clicking “Apply online” below. The application must include the following documents/attachments:
Motivated letter of application
Curriculum vitae incl. education, experience, previous employments, language skills and other relevant skills
Certified copy of diplomas/degree certificate(s)
Certified copy of transcript of records
Letter of recommendation
In all cases, ability to perform the job will be the primary consideration, and thus we encourage all – regardless of their personal background and status – to apply.
Application deadline: 15 November 2017
For further information, please contact Professor Henrik Semb by e-mail semb@sund.ku.dk
Send your application with your CV as well as names and contact details of referees electronically by clicking the ´Apply online´ below. We only accept electronic applications.
During spermatogenesis, progenitor cells must undergo tightly regulated changes to produce functional gametes. However, the genetic control of this process in humans has eluded researchers. This week we feature a paper published in the latest issue of Development that describes the changing genetic expression of cell during spermatogenesis. The co-first authors Sabrina Jan and Tinke Vormer and PIs Sjoerd Repping and Ans van Pelt of The University of Amsterdam told us more.
Sjoerd Repping, Tinke Vormer, Sabrina Jan and Ans MM van Pelt
Sjoerd and Ans, can you each give us your scientific biographies and the main questions your lab is trying to answer?
SR & AvP We work at the Center for Reproductive Medicine of the Academic Medical Center of the University of Amsterdam. Our center focusses on providing top-clinical care to patients suffering from infertility and on understanding the basic pathophysiological mechanisms underlying infertility. Our research laboratory has four active lines of research: 1- Spermatogenesis and spermatogonial stem cells, 2- Preimplantation embryo development, 3- Genomic stability of reproductive cells, and 4- Placentation and gestational diseases. Besides our laboratory research, our department is very active in healthcare evaluation research where we try to increase effectiveness and safety of medically assisted reproduction.
Sjoerd was trained as a clinical embryologist, received his PhD on genetics of male infertility at the University of Amsterdam and the Whitehead Institute in Cambridge and is currently professor of Human Reproductive Biology, head of the Center for Reproductive Medicine and Director of the Amsterdam Reproduction & Development Research Institute.
Ans was trained as a stem cell biologist. She received her PhD on the role of vitamin A on spermatogenesis and spermatogonial stem cells at the Medical School of the Utrecht University in collaboration with the Hubrecht Institute, both in the Netherlands. She is currently associate professor at the Center of Reproductive Medicine, head of the Reproductive Biology Laboratory and leader of the research line Spermatogenesis and spermatogonial stem cells.
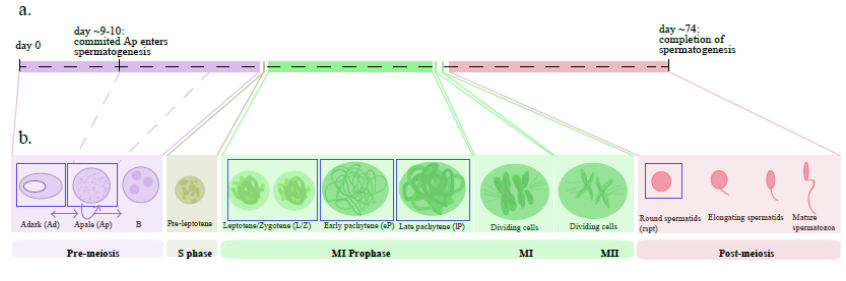
Timeline of spermatogenesis, from Figure 1, Jan, Vormer et al, 2017
Sabrina and Tinke, how did you both end up working on this project?
SJ I started working on this project as part of my PhD training. I have always been very interested in the field of reproductive medicine. In 2008 I completed a masters degree in reproductive and developmental biology. In 2011 I was looking to further academic training and as soon as I read this project’s research proposal I was immediately interested. For me, spermatogenesis is a unique process from which we can learn so much not only about gamete formation and fertility but also about the molecular control of processes such as stem cell biology, mitosis, meiosis and cytodifferentiation.
TV Before joining the Centre for Reproductive Medicine, I performed my PhD studies at the Netherlands Cancer Institute in Amsterdam, where I studied the molecular changes that occur when a healthy cell transforms into a cancer cell. As such, the step to studying the molecular changes during spermatogenesis is not as big as people sometimes think. During my undergraduate studies, I performed a literature study about molecular changes during spermatogenesis and I was absolutely amazed by this process. After finishing my PhD studies, I saw an advertisement for a postdoctoral position in the Centre for Reproductive Medicine, and immediately applied.
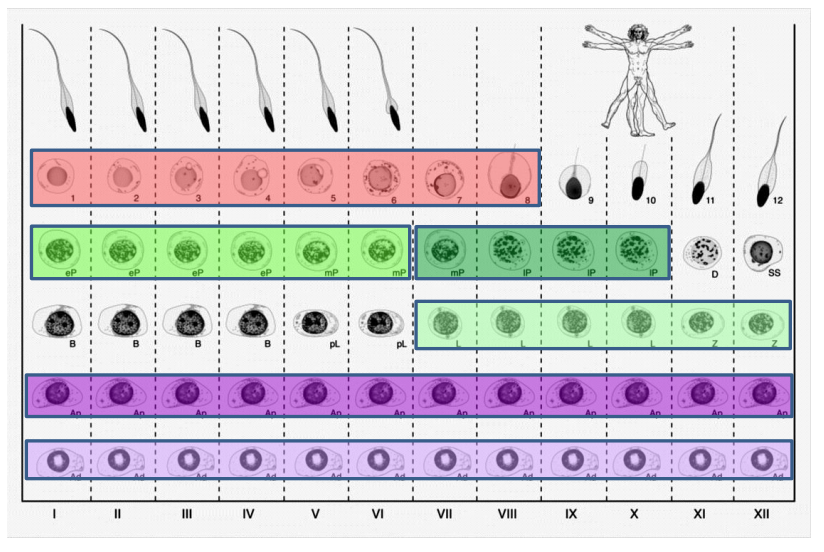
The stages of the seminiferous epithelium in man, from Figure 1, Jan, Vormer et al, 2017
What was known about the molecular control of spermatogenesis in humans prior to your paper?
AvP The molecular control of human spermatogenesis was largely unknown. Only recently a few groups in the world have investigated the transcriptome and epigenetic patterns of the entire human testis or some isolated testicular cell types, but not to the extent as we have now been able to do.
Can you give us the key results of the paper in a paragraph?
AvP & SJ In this paper we investigated the transcriptome of male germ cells using laser capture microdissection to isolate testicular cells based on morphology and localization in the testis. By doing so, we were able to identify the RNA profiles of many more subpopulations of germ cells than with any other method described before. This resulted in a far more detailed understanding of the molecular regulation of human spermatogenesis.
TV We identified the onset of major gene expression changes during human spermatogenesis and discovered the specific timing of transcriptional changes of individual genes. Also, we found that Adark (quiescent), Apale (actively dividing) spermatogonia display similar transcriptomic patterns and that the transcriptome of precursor cells already contains genes necessary for cellular differentiation later in development.
Can you briefly describe the technique you developed and how it could be used to answer other biological questions?
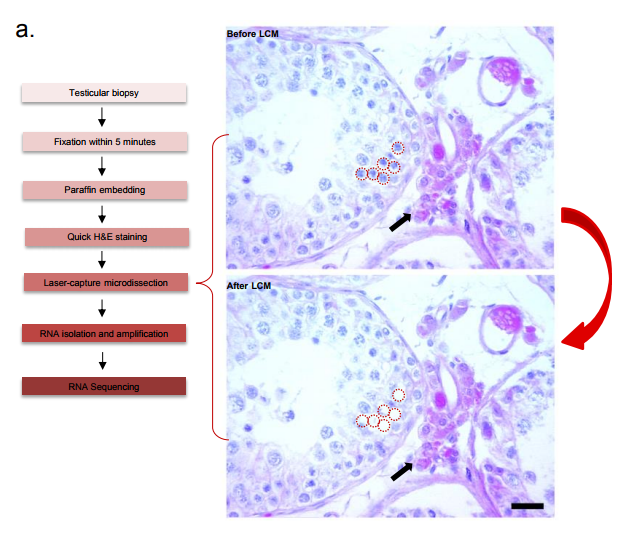
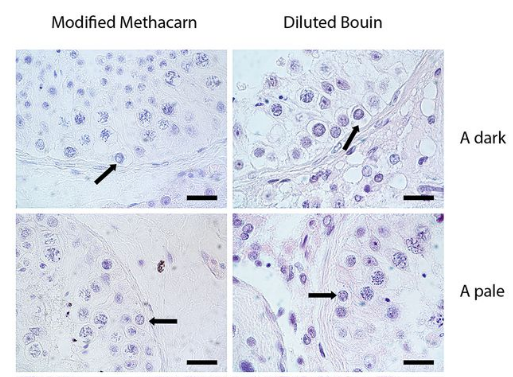
AvP & TV The technique is based on single cell laser capture microscopy out of fixed and paraffin embedded tissue. During this project, we learned that the fixative is of crucial importance. Not only for the recognition of various cell subtypes in the tissue based on their nuclear structure, but also to maintain RNA integrity to allow for in-depth transcriptome analyses of the isolated cells. In our case with testis tissue, we ended up with an alcohol-based fixative instead of the classically used Bouin’s fixative. This alcohol-based fixative maintains nuclear structure and RNA integrity and can be applied to any other tissue to allow for single cell capture. By using laser capture microscopy, we were able to isolate pools of single human germ cells from these alcohol-fixed specimens. This enabled us to analyse the transcriptome of human germ cells during the different stages of spermatogenesis. The technique can be used to study other tissues and developmental processes of interest.
Technique protocol to obtain samples for sequencing, from Supplementary Figure 2, Jan, Vormer et al, 2017
How will your technique help future research into male infertility and possible treatments?
AvP We can now use the results of the current paper as a reference for further transcriptome analyses of germ cells from patients with a maturation arrest in spermatogenesis. In doing so, we will gather information on the molecular aberrations underlying male infertility, which will give us detailed insights for establishing future treatment strategies.
When doing the research, did you have any particular result or eureka moment that has stuck with you?
SJ A moment that will always stick with me while working on this study is the moment I saw the first sequencing results. Many had doubted if this project would be feasible considering the small amounts of RNA we were attempting to experiment with, and it indeed took us years to be able to do so. It is astonishing how much one can do with extremely small amounts of RNA (picograms) (and will power!); we were able to generate whole transcriptomic profiles. This gives new meaning to the phrase “nothing is impossible”.
TV When setting up the technique, we had to find the correct fixative, microscope settings and a reliable RNA amplification method to be able to analyse our cells using next generation sequencing. Once this all worked out and we got our first sequencing runs: great!
The morphologies of the the germ cell subtypes in testes tissue using two different fixatives from Figure 2, Jan, Vormer et al, 2017
And what about the flipside: any moments of frustration or despair?
SJ There were certainly moments of frustration but certainly not despair. We had a period during the project in which the tubes used for laser capture microdissection were malfunctioning resulting in loss of our carefully collected germ cells. Capturing spermatogonia (germ cells that require careful microscopic evaluation) takes a long time – on a good day we captured 50 germ cells in 3-4 hours. So, you can imagine that loss of such precious collections causes a lot of frustration. Thankfully, it was a batch problem and we could easily solve this problem with the tube suppliers.
TV The isolation of single cells under the microscope is a concentrated job. Especially because we had to set up the technique, this involved quite some patience. Once an experiment failed after carefully isolating all the individual cells, this could be a very frustrating moment… Luckily, Sabrina and I could support each other!
What are your career plans following this work?
SJ At this moment I am in training to become a molecular clinical geneticist at the University Medical Center Groningen in The Netherlands. This project was inspirational in my current career path. Working on spermatogenesis, I was able to work on RNA expression but was also able to gain knowledge on the role genetics plays in the pathophysiology of spermatogenesis. This sparked my interest in genetics. Since RNA expression is a growing field in genetics, in my current work I get to merge these two very interesting worlds together.
TV I am currently working as a medical science liaison for a pharmaceutical company. The research for this paper was performed in close collaboration with physicians that treat infertile patients. I found this very inspiring and hope that our work will contribute to the development of future treatment strategies. In my new role, I am close to the development and application of new therapeutics in the clinic and have regular contact with physicians, which I find very rewarding.
What’s planned next for the Repping and van Pelt labs?
SR & AP One of the most exciting things we are working on is bringing autotransplantation of spermatogonial stem cells (SSCs) into clinical practise for sterile childhood cancer survivors. Often children with cancer become sterile due to the chemotherapy they receive to treat their cancer. We have pioneered the development of an in vitro culture system of human SSCs and are currently in the progress of finalizing crucial in vitro and animal safety studies before we embark on the first clinical trial in humans. Furthermore and in line with the current study, we aim to study the molecular aberrations in men suffering from idiopathic infertility. The ultimate goal there would be to treat these men with a therapy, perhaps in combination with SSC autotransplantation, that will allow these men to produce sperm again and become a genetic parent.
Lastly, what do each of you like to do when you’re not in the lab?
SR I enjoy taking long distance runs (just ran my 6th marathon), reading and spending time with my family and friends.
AvP I enjoy hiking, cycling and spending time with my family and friends.
SJ I enjoy going out dancing with my friends, swimming and various other activities such as walking in the various different beautiful dunes in The Netherlands and last but certainly not least, spending time with my family.
TV I am a big fan of flamenco dancing! I have been doing that for quite a few years, and I recently started with a flamenco-singing class for dancers. It is a lot of fun, I really enjoy the musicality and temperament of this music. Of course I also enjoy spending time with family and friends.
Sabrina Z. Jan, Tinke L. Vormer, Aldo Jongejan, Michael D. Röling, Sherman J. Silber, Dirk G. de Rooij, Geert Hamer, Sjoerd Repping, Ans M. M. van Pelt. 2017. Unraveling transcriptome dynamics in human spermatogenesis.Development. Volume 144, Issue 20, p3659-3673.
This is #30 in our interview series. Browse the archive here.
The CPH Bioscience PhD programme is designed for international talents to come to Denmark and start their research careers at one of the NNF Research Centers.
The Copenhagen Bioscience PhD programme recruits up to 16 motivated international students annually to launch their careers in the vibrant scientific environment of the Novo Nordisk Foundation Research Centers in Copenhagen. For enrollment in September 2018, applications is now open until December 2017.
Selection is based on academic achievements, research experience, academic references and interviews. A mandatory interview visit for up to 40 shortlisted applicants comprises panel interviews, one-on-one meetings with potential supervisors, and tours of the four Novo Nordisk Foundation Research Centers, and will take place in Copenhagen in March 2018. The Novo Nordisk Foundation will pay for travel and accommodation for selected applicants in association with the interview visit.
The University of Manchester, 2018/19 BBSRC DTP PhD Project
Understanding tubulin regulation during neuronal development, ageing and degeneration
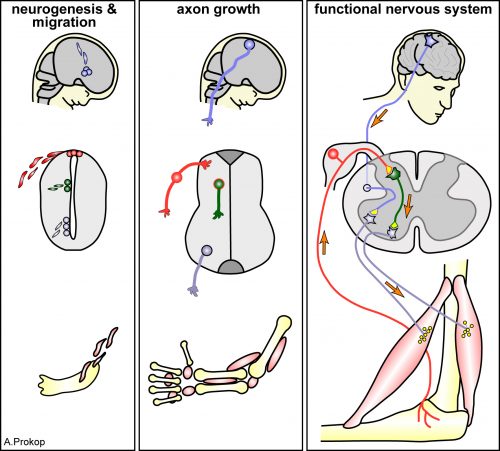
Axons are slender, up-to-a-meter long, cable-like extensions of neurons which form the nerves and nerve tracts that wire our bodies and brain. These delicate cellular structures have to be maintained for an organism’s life time and are often the first to be affected in ageing, injury and neurodegeneration. To understand such conditions and identify ways to improve axon maintenance and regeneration, we study the regulation of microtubules (MTs) which form parallel bundles running all along axons to form their structural backbones and life-sustaining transport highways.
Axons are the cables that wire the nervous system (LINK)
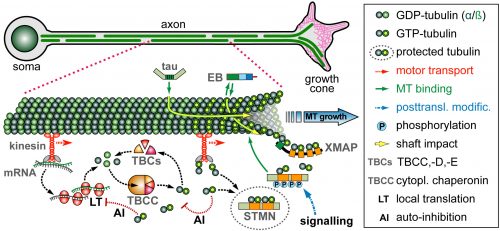
On this project, you will study how the polymerisation of MTs within these bundles is regulated to drive axon growth and prevent senescence during ageing. The key question is how tubulins (i.e. the building blocks of MTs) are made available and continuously supplied in the narrow axons, up to a meter away from the cell body. This fascinating topic is most relevant to axon biology and pathology but surprisingly little understood. Your pioneering work will be based on our recently published mechanistic model of axonal tubulin supply and MT polymerisation, deduced from our own data and general knowledge in the field [Ref.1]. You will use cutting edge methodologies to study (1) contributions made by axonal transport and local tubulin translation, (2) roles of the chaperone machinery of tubulin assembly, and (3) mechanisms of tubulin storage and gene expression regulation.
Proposed model of tubulin provision: Tubulins in axons require long distance transport, assembly via chaperones, STMN-mediated storage protecting them from auto-inhibition of their own biosynthesis (LINK)
For your studies, you will use neurons of the fruit fly Drosophila, which provide uniquely powerful genetic and cell biological means in order to efficiently generate new understanding that can then be applied to higher animals [Ref.2; LINK]. You will be able to capitalise on expertises of the supervisory team: the host group (Andreas Prokop) has long-standing experience with MT regulation and the Drosophila neuron model, the first co-supervisor’s group (Thomas Waigh) with high resolution imaging and quantitative approaches [Ref.3; Ref.4], and the second co-supervisor’s group (Mark Ashe) with RNA visualisation and processing [Ref.5].
Cultured Drosophila neurons carrying tubulin mutations (middle) or STMN mutations (right) grow shorter than normal neurons (left)
Your transferable and experimental skill training will include genetics, cell biology, molecular biology, imaging techniques (live, high resolution, axonal transport, spatial detection of RNA and translational activity), quantitative analyses and modelling, as well as expertise in the important research areas of cytoskeleton and neurobiology. Finally, Andreas Prokop is an expert in science communication [Ref.6] providing further training opportunities important for your future career.
Deadline for applications is Fri 2nd of Nov. 2017, 5pm
Interviews are held on Tue/Wed, 9th/10th Jan. 2018
Offers will be confirmed in mid-Feb. 2018
References
[1] Voelzmann, A., Hahn, I., Pearce, S., Sánchez-Soriano, N. P., Prokop, A. (2016). A conceptual view at microtubule plus end dynamics in neuronal axons. Brain Res Bulletin126, 226-37
[2] Prokop, A., Beaven, R., Qu, Y., Sánchez-Soriano, N. (2013). Using fly genetics to dissect the cytoskeletal machinery of neurons during axonal growth and maintenance. J. Cell Sci.126, 2331-41
[3] Kenwright, D.A., Harrison, A.W., Waigh, T.A., Woodman, P.G., Allan, V.J. (2012) First-passage-probability of active transport in live cells, Physical Review E, 86, 031910
[4] Georgiades, P., Allan, V.J., Dickinson, M., Waigh, T.A. (2016) Reduction of coherent artefacts in super-resolution fluorescence localisation microscopy, Journal of Microscopy, 264, 3, 375-383
[5] Sfakianos, A. P., Whitmarsh, A. J., Ashe, M. P. (2016). Ribonucleoprotein bodies are phased in. Biochemical Society Transactions44, 1411-1416
[6] Illingworth, S., Prokop, A. (2017). Science communication in the field of fundamental biomedical research
Recommended by Guojun Sheng, IRCMS, Kumamoto University, Japan.
Mammals alive today are split into three groups: the placentals (like us), the marsupials (like Skippy), and, distantly related to the rest, the monotremes. Monotremes come in two kinds: echidnas, “shambling, animated pincushions” whose young are known as ‘puggles’, and platypuses, famous for their chimeric collection of weird anatomical features (their species name, Ornithorhyncus anatinus, means ‘duck-like bird-snout’).
In addition to their odd appearance, monotremes stand out compared to other mammals when it comes to reproduction: rather than giving birth to live young, they lay eggs, a trait shared with birds and reptiles, which together form the sister group to mammals (mammals, birds and reptiles together form the amniotes). Given their phylogenetic position and mix of biological traits, monotremes might tell us a lot about mammalian evolution from a reptile-like ancestor, keeping in mind of course that they have not stayed still since they split from the rest of the mammals perhaps a hundred and sixty million years ago. From the perspective of developmental biology, the question is how an ancestral, reptile-like mode of embryogenesis was transformed into a mammal-like one: do monotremes develop more like other mammals, or more like birds and reptiles? (Another proviso here: ‘mammal-like’ rather obscures the diversity seen in early mammalian embryogenesis, as explored by Guojun Sheng, Marilyn Renfree, Berenika Plusa and others, but monotremes might in fact prove a useful example to understand this diversity.)
Answering these questions is not easy. Some animals are more amenable to embryological investigation than others, and monotremes are exceptionally intransigent in this regard: they can be hard to find, do not breed well in colonies (though this might be getting easier), are not really prodigious breeders in any case, and have embryos that are difficult to collect and preserve. ‘Non-model’ doesn’t quite cover it.
Nevertheless, monotreme embryology has a long history. The Scottish zoologist W.H. Caldwell was one of first Western scientists to unequivocally describe monotreme egg-laying (read more in Brian Hall’s paradoxical history of the platypus), in a famously terse telegram sent while on an expedition in Queensland in 1884 :
“Monotremes oviparous, ovum meroblastic”
‘Meroblastic’ describes the incomplete cleavage of the early embryo, which reptiles and birds do too.
Plate from Caldwell’s The Embryology of Monotremata and Marsupialia Part I, available at The Embryo Project
Caldwell’s expeditions were torrid affairs –
“Crossing the Maclntyre River in a flood, the buggy was upset, and its contents washed away. The two following months were lost through the effects of a fever…”
But not just for him: the Aboriginal Australians employed by Caldwell to catch the animals were treated horrendously, according to the introduction to his paper describing the expedition (it’s quite a shocking read, but perhaps not an altogether surprising one).
As Caldwell describes, collecting monotreme samples in the wild was a difficult and often brutal business. Since a lot of embryonic development occurs in utero, to get earlier stages you have to kill the mothers. Platypuses posed particular problems, as described in a later account:
“…it is extremely difficult, even for residents in Australia, to procure material necessary for an investigation into the development of the egg of Ornithorhynchus….The animal itself, though pretty widely distributed, and probably still far from becoming extinct, is to be found, in any one locality, only in comparatively small numbers.
The eggs, when laid, are deposited in a burrow which it is far from easy to locate, and whose opening up involves a considerable amount of labour, since, apart from its great length, the river-bank in which it is situated is commonly enough permeated by tree-roots. And when at length the actual dwelling-chamber or nest is successfully opened up, no reward at all may be forthcoming, or the material which is obtained may be unsuitable for the immediate purpose in view.
Even when it is the intra-uterine stages of the egg which are required…the difficulties are nearly as great. The animal is extremely shy and difficult [to] approach. They are occasionally, but rarely, captured as an incident in net-fishing in the larger rivers: otherwise they are practically only obtainable with the gun. During the breeding season, however, the pregnant female appears to keep much more closely to the burrow, so that one may then commonly enough shoot five or six males to one female.”
This was published in 1908 by J.T Wilson and one J.P. Hill, who with T.T. Flynn wrote the two hefty monographs that are the subject of this Forgotten Classics piece – Volume IV and Volume VI of the Transactions of the Zoological Society of London’s Development of the Monotrema series.
Hill was born in Edinburgh but moved early in his career to Sydney (where he met Flynn who was studying there), before returning to the UK to University College London and rising to chair of embryology and histology. Flynn was an Australian who moved to Tasmania from Sydney to become a professor of biology and an eminent naturalist. In Tasmania, as well as collecting monotreme samples, he fathered Errol Flynn (yes that Errol Flynn, the actor), but after his funding was cut he moved to Queens University in Belfast. There he served as a fire martial and casualty clearing officer in the Second World War, for which he was awarded an MBE.
The two monographs describe observations on a collection of 150 or so monotreme eggs and embryos initiated in 1896 by Hill in Queensland, and later added to by Flynn in Tasmania. The samples were preserved in the field and then sectioned, stained and analysed in the lab, often decades later (one was embedded in wax thirty years before being sectioned with a myotome; there may have been half a century between a sample’s collection and the publication of its description). Volume IV describes the making of the monotreme oocyte, its fertilisation and the early cleavage stages of the embryo. Volume VI goes from cleavage until germ layer formation.
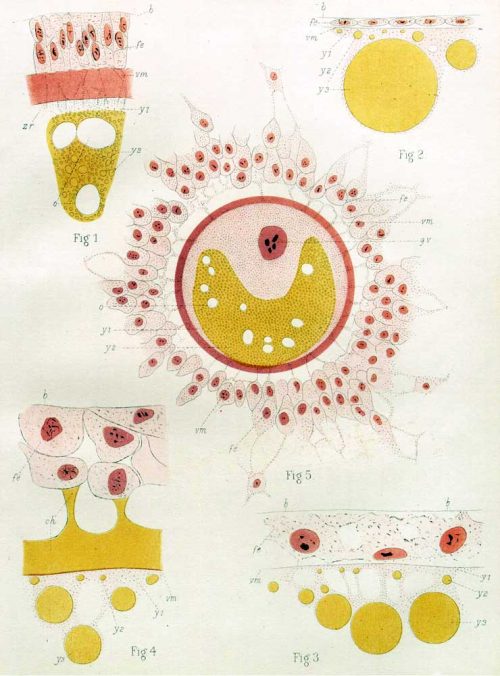
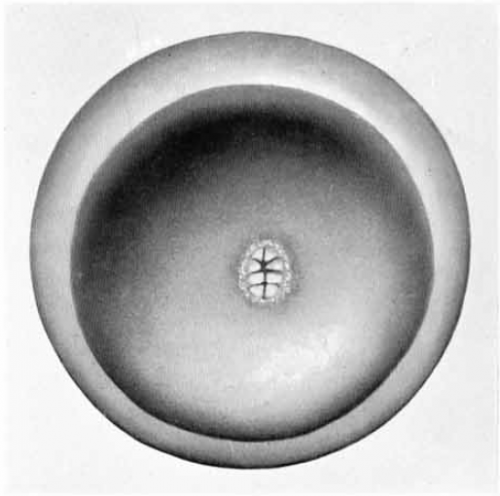
A platypus egg, Plate 91 from Flynn and Hill, 1939, with permission of Wiley.
The monographs are methodical and descriptions of the collection – each sample is described in the most minute of details, consistent with Hill’s renown for accuracy both in sample preparation and description (“everything he does is done with the most meticulous accuracy…the exact wording of each sentence had to be discussed to make certain that it was precise, unambiguous, and made all possible reservations”, according to the introduction to a dedicated volume of the Journal of Anatomy). It can make for some daunting reading, particularly if you are unfamiliar with some of descriptive terms – one section on the oocyte features eosinophil and basal granules, pseudo-reticular strands, vitello-fatty zones, irregular sinuous folds, linin threads. But in fact much of it is written beautifully and the plates are often quite stunning.
Playpus oocyte. Figure 34, Plate VII, Flynn and Hill, 1939.
Echidna, section through the follicular wall. (f.s. = follicular secretion ; zp = zona pellucida; th.e. & th.i., = theca externa and interna; f.e. = follicular epithelium). Fig. 70, Plate XIV, Flynn and Hill, 2939.
Echidna. Section of germinal disc, showing the male and female pronuclei overlapping each other at the centre of the formative zone of the disc (fcd). (vcd. = deep vacuolated zone of the disc; mg.x. = marginal zone; shm. = shell-menibram; yb. = yolk-bed; zpa. = zona-albumen layer.) Fig. 77, Plate XV, Flynn and Hill, 1939.
Echidna, showing male and female pronuclei together. Figure 81, Plate XV, Flynn and Hill, 1939.
Platypus, 8 cell stage, showing mitosis. Figure 96, Plate XVIII, Flynn and Hill, 1939.
Echidna blastodisc, transverse view. (gr. = germ-ring; pv. and smv. = peripheral and sub-marginal vitellocytes). Fig. 56, Plate IX, Flynn and Hill, 1947.
Echidna blastodisc, surface view. Fig. 67, Plate XI, Flynn and Hill, 1947.
Echidna, showing a portion of the unilaminar blastodermic membrane (emc = endodermal mother-cell; p.ect. = prospective ectoderm cell). Fig. 99, Plate XVII, Flynn and Hill, 1947.
So how do you make a monotreme? Flynn and Hill observed that many features of monotreme oocyte formation and embryogenesis look quite like their bird and reptile cousins. For instance the eggs are yolky, the germinal disc sits on top of the yolk, and early cleavages are meroblastic; early cleavage creates a cellular blastoderm that is separate from the uncleaved yolk; there is an extra-embryonic structure called a germ-ring not seen in mammalian embryos; the multi-layered blastoderm thins out into a single layered structure, and this process appears to involve migration of cells to within the embryo, which looks like reptile delamination. There are also some features that are more mammal-like, and some which seemed to Flynn and Hill to be unique to monotremes, but the key observations seem to be that these mammals share a lot of features ofearly embryogenesis with birds and reptiles.
As discussed by Guojun Sheng below, Flynn and Hill’s findings suggest we should do more to understand the early development of birds and reptiles, stages which have historically, and for technical reasons, been underserved. We could then move to a more complete understanding of the variations of amniote development, and hence its evolution.
The work also makes us consider the use of different models in developmental biology. There are many animals for which descriptive embryology and anatomy of the kind practised by Flynn and Hill was the only way in, and for which today perhaps only genome sequences – the platypus genome was sequenced in 2007 – can add to our understanding. When such animals are the only extant outcomes of critical evolutionary junctures, papers like Flynn and Hill’s are crucial. And, of course, we are benefiting from work that was of its time: such a collection with its collateral damage of thousands of dead monotremes simply could not have been assembled today. No one has performed a similar analysis since, so the papers really do stand alone in embryology.
These two related, heavy-weight (literally) papers by the same authors (TT Flynn and JP Hill) [for biographical accounts, see http://adb.anu.edu.au/biography/hill-james-peter-6669 and http://adb.anu.edu.au/biography/flynn-theodore-thomson-6202 ] describe pre-gastrulation development of monotreme embryos. Despite the length (each over 150 pages), these two papers are easy and enjoyable to read. The clarity and quality in their writing style and data presentation can only be appreciated by reading the original papers, rather than from summaries in a handful of review papers which cited them.
What makes them essential readings for developmental biologists is their relevance to modern-day stem cell biology, the foundation of which is based on our understanding of mammalian early development. The monotremes are a group of prototherians (early-branched out mammals) which retain many developmental features of their reptilian ancestor. Our knowledge on cellular and molecular regulation of early lineage segregation (trophoblast, epiblast and hypoblast) in eutherians (placental mammals including mice and humans) would be incomplete, to say the least, without an understanding of these events from the perspective of comparative mammalian/amniote embryology. Although experimentation with monotreme embryos is impractical, similarities between monotreme and reptilian (including avian) early development make a strong case for redoubling our efforts in the investigation of pre-gastrulation development using avian/reptilian models.
Readers may be interested in two papers from our lab:
1) Research paper on chicken pre-ovipositional development (published in Development)
Both papers have been made free to view for three months courtesy of Wiley Publishers.
Aidan Maartens
This post is part of a series on forgotten classics of developmental biology. You can read the introduction to the series here and read other posts in this series here. We also would love to hear suggestions for future Forgotten Classics – let us know in the comments box.
Supervisors: Dr Jill Harrison and Dr Martin Homer.
Plant shapes range from tiny string or mat-like forms to massive multilayered upright forms with complex organ systems such as shoots, roots and leaves. Despite these wide differences in shape, many plant gene families are very ancient, predating shape diversification. We can therefore study mechanisms for shape determination in simple plants such as liverworts, and use the knowledge gained to understand plant shape determination in general.
To this end, we previously used a combination of live imaging, statistical model fitting, computational modelling and molecular biology to discover mechanisms regulating shape in the liverwort Marchantia polymorpha (Solly et al. (2017): Current Biology).
We found that Marchantia undergoes a stereotypical sequence of shape transitions during development. The overall shape depends on regional growth rate differences that are specified by the growing apical notches. Computational modelling showed that a diffusible, growth-promoting cue produced in the notches is likely to pattern these regional growth rate differences, and pharmacological experiments suggested that the plant hormone auxin equates to this growth-promoting cue.
New models suggest a role for differential oriented growth (anisotropy) in Marchantia shape determination. Anisotropy emerges as an outcome of underlying tissue polarities, and directional auxin transport may have a role.
Your project will build on the work above to determine how auxin contributes to plant shape determination in Marchantia.
Training:
By combining computational and wet lab approaches, your project work will provide training at the cutting edge of the plant evolution and development fields. You will benefit from further formal teaching and internships included in the SWBioDTP programme. The skills and techniques you learn will be broadly applicable in the academic biology and biotech sectors and widely transferable amongst areas such as science policy, publishing and computing.
Reading:
Harrison (2017). Development and genetics in the evolution of plant body plans. Philosophical Transactions of the Royal Society B372: 20150490.
Hong and Roeder (2017). Plant development: differential growth rates in distinct zones shape and ancient plant form. Current Biology27: R19-21.
Solly et al. (2017). Regional growth rate differences specified by apical notch activities regulate liverwort thallus shape. Current Biology27: 16-26.
Whitewoods and Coen (2017). Growth and development of three-dimensional plant form. Current Biology27: R910-918.
Further information:
The deadline for applications is 4th December 2017. Please contact Dr Jill Harrison (jill.harrison@bristol.ac.uk) for informal discussions about the project. Further information about project supervisors’ work can be found on Jill Harrisonand Martin Homers’ home pages. Further information about the SWBioDTP and application procedures is listed on the SWBioDTP web pages.
Supervisors: Dr David Ferrier (University of St Andrews) and Prof Kate Storey (University of Dundee)
This project will dissect the regulatory mechanisms of the chordate ParaHox genes (Gsx, Xlox/Pdx1 and Cdx), analysing regulatory elements of these genes in both the invertebrate sea squirt Ciona intestinalis and the vertebrate Gallus gallus (chicken). ParaHox genes are the evolutionary sisters to the Hox genes, and like their sisters are important components of axial patterning, mainly in the central nervous system and gut. They also tend to have a clustered organisation in the genome that is likely linked to how the genes are regulated. Mis-regulation of ParaHox genes can cause diseases such as diabetes and colon cancer, and changes to the Hox/ParaHox genes are important agents in the evolution of animal form.
In this project, we will capitalize on the power of the comparative approach to deduce underlying fundamental aspects of body axis patterning by regulation of the ParaHox genes.
The student will obtain training in cutting-edge techniques in molecular biology, embryology, bioimaging and bioinformatics and be part of the enthusiastic and vibrant research communities in the Universities of St Andrews and Dundee, benefitting from the complementary strengths, strong links and close proximity of these institutions.
Funding Notes
Applications for BBSRC EASTBIO studentships are invited from excellent UK students (and EU citizens if they meet UK Research Council residency criteria) with at least a BSc (Hons) 2.1 undergraduate degree.
 (No Ratings Yet)
(No Ratings Yet)
 (1 votes)
(1 votes)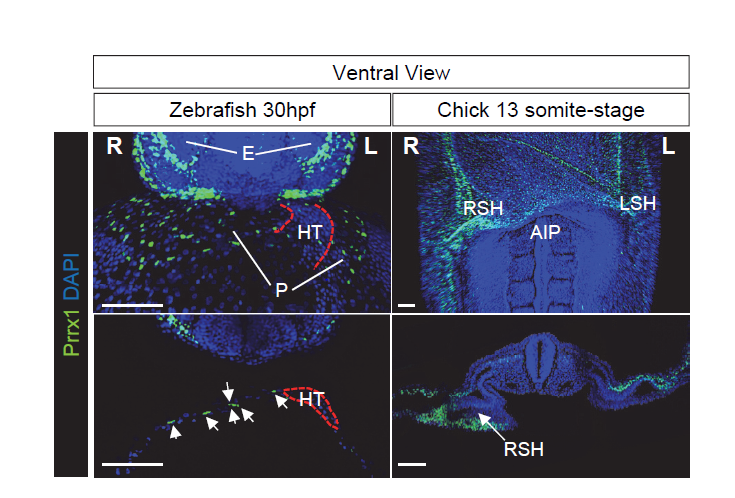











 (8 votes)
(8 votes)