Innovations in science are happening all over the nation! Visit the science videos at the 2017 STEM for All Video Showcase, funded by the National Science Foundation. Interact with the community by asking questions, getting answers, and voting for your favorite video! Support STEM education and let your voice be heard from May 15-22 at the STEM for All Video Showcase!
Your vote counts!
Did you see an amazing video at the showcase? Let us know! You can vote for your favorite video through Twitter or the showcase website. The video with the most votes will be given the “Public Choice” award on Tuesday, May 23, and the winning participants will be recognized by the National Science Foundation!
Do you support STEM? Let the world know!
You make the difference. Please share the STEM for All Video Showcase with your friends and colleagues, on social media, and with your local science centers. Help us spread the word about the amazing people working in STEM education and the awesome work they are doing for our communities!
More details on the event:
The Stem for All Video Showcase is funded by the National Science Foundation. This year’s theme is “Research & Design for Impact.” In short 3 minutes videos, leaders of STEM education initiatives describe how their projects are meeting some of today’s most pressing scientific and technological challenges. The STEM for All Showcase is a collaborative effort of the following NSF resource centers: MSPnet, CADRE, CAISE, CIRCL, STELAR, CS For All Teachers. It is funded by the National Science Foundation (#1642187).
The Showcase is powered by the Videohall.com platform developed by TERC, a STEM-focused education research nonprofit based in Cambridge, MA.
I had started to become a little worried when I didn’t see Eric on the opening day of the conference, but it turned out that his plane to Germany had been delayed by the snowstorms blanketing the Eastern seaboard of the US and he made it in the end. Between sessions later on, we found a slightly chilly empty computer lab to sit down in, and over the course of an hour talked about his history in research and views on science in general. My lasting impression was of an enthusiasm and passion for research that was by equal measure refreshing and infectious – it made me want to look down a microscope again!
Eric Wieschaus is a HHMI Investigator, and the Squibb Professor in Molecular Biology at Princeton University and the Lewis-Sigler Institute for Integrative Genomics. In 1995, he was awarded the Nobel Prize jointly with Edward Lewis and Christiane Nüsslein-Volhard for discoveries about the genetic control of Drosophila embryogenesis. We caught up with Eric at the joint meeting of the German and Japanese Societies of Developmental Biologists held in Kiel in March 2017, and discussed his career, his thoughts on the field and the impact the Nobel award had on his life.
Eric in the lab
I understand that after your first experience with Drosophila – as an undergraduate in Harvey Bender’s lab at Notre Dame – you said never wanted to see a fly again! What led you to change your mind?
I suppose the formative undergraduate experience wasn’t washing dirty fly bottles and making fly food, which is what I had done in Harvey Bender’s lab, but an embryology course where we looked at frog and chick embryos, which were the embryos people worked with at the time. I knew then that I wanted to be an embryologist, to study development. But I think I just didn’t know that flies had embryos! Anyway, I ended up at Yale in New Haven, and for various reasons I was assigned to the lab of Donald Poulson. Poulson had, in the 1930s and 1940s, first as a graduate student and through to his professorship, described all of Drosophila embryogenesis. So I knew that I wanted to study embryos, and I had by chance landed in the laboratory of the one person in the world who really knew how Drosophila embryos develop. Poulson was a very kind person: he took me in to his lab when he specifically didn’t want to have a graduate student. It took him a little while to find a young assistant professor who was willing to take me on, and that turned out to be Walter Gehring.
And as a graduate student with Gehring, you were trying to map cell lineage in Drosophila with somatic clones and disc transplantation. What was the drive behind this work?
Walter had developed a tool to culture embryos, based on the strategies people had used to culture imaginal discs. He was able to convince me, a naïve young graduate student entering the lab, that the best experiment in the world would be to isolate a single cell from the blastoderm, grow it in these culture conditions and measure its developmental capacity, to establish whether it had already undergone some determination or programming. And that was my project – I spent my first three years grinding up embryos and trying to get single cells to grow. I have to say that none of that ever worked, but at some point I decided that I needed to have a control – once I had these wonderful cultures going, and these cells displaying their inherent potential, I wanted to be able to compare them to what a blastoderm cell actually did if left in situ. I decided I would produce clones at the blastoderm stage and follow those cells, but I never really looked at them because I still believed my experiments would work. But at the end of four and a half years, when I realised that I wasn’t going to have cultured cells as part of my thesis, I decided to go back and look at the controls. Luckily, it turned out that what cells normally do in development is almost as interesting as what you can get them to do when you artificially manipulate them.
When Walter’s lab moved to Basel, you were brought into contact with Christiane (Janni) Nüsslein-Volhard. How did that work out?
Janni and I both finished our thesis work at around the same time – she was in Tübingen, and having done molecular biology was an attractive person for Walter to bring to his lab. She actually came to Basel specifically not to do molecular biology, but instead to learn how to look at embryos, and as I was the only person in the lab who was working with embryos, we bonded over that in the four months before I left. And later, we both were fortunate to get positions in Heidelberg – these were real group leader positions, but we didn’t know that we had to bargain for space! So we ended up together, sharing a lab, and we already knew kind of what we wanted to do; it was a perfect set of circumstances.
And in Heidelberg, you began work on the screen that would eventually win you the Nobel Prize. Screens obviously require a lot of time and effort: how did you stay focussed and cope with the practical demands at the time?
Well it was a lot of work, but the payoffs were real. We spent a certain amount of time figuring out how to do the screen, and that was maybe more frustrating than actually doing it. But once we started the big screen, scaled up and lasting about two and a half months, we discovered something new every day, and so focus was not at all hard. We would work from nine in the morning to around midnight, pretty much every day of the week, but the payoff was already there – not in understanding as such, but in the realisation that we were seeing stuff that no one had ever seen before, and thinking thoughts that no one had ever thought before. I don’t even remember being frustrated or wanting to slow down at all – my memory of that time is just of the fascination and excitement of doing it.
We were seeing stuff that no one had ever seen before, and thinking thoughts that no one had ever thought before
Was there a key lesson the screen taught you about how development works?
Before we did the screen, we really didn’t know what we would get out of it. The two dangers were that every gene that you mutated would mess up development, and that the phenotypes would be complicated and heterogeneous and impossible to interpret. But what actually happened was that the number of zygotically active genes we found was small – about 120 to 130 – and the mutants had unique phenotypes that identified specific processes, which meant you could immediately group things. This small number contrasted with the characterisation of RNA heterogeneity carried out at the time, which estimated there were thousands of different mRNAs present in the embryo, but most of these turned out to come from the mother. So the conceptual lesson was that the embryo uses transcription of a small number of genes to drive specific events forward. If gene products can be supplied to all cells, they’re provided maternally, but if you want to turn the gene on at a specific place or at a specific time, zygotic transcription does that. That was the power of the screen, to pick up these regulators: the genes controlling developmental decisions. It seems quite obvious now, but did I predict that at the time? Absolutely not.
Following the screen, you moved your lab to Princeton in 1981. What did you hope to achieve in the early days in New Jersey?
Initially, when we returned to Princeton, I helped Trudi (Schüpbach) a bit when she was setting up her maternal effect screen to find the determinants deposited in the egg by the mother. But what I really wanted to understand was morphology, and this very quickly became a cell biological problem, of how cells move and change shape and so on. This obviously required a lot of observation, and I remember at the time the great discovery was that there was something called phalloidin (which labels F-actin) – it was fluorescent, and you could buy it from a supplier! You could get a fluorescence microscope and just look – it really allowed you to see embryogenesis differently. So this period was also a time where, as well as advances in molecular biology, there were great advances in cell biological reagents, which you could begin to apply to embryos to think about these processes from a more cell biological perspective. It all came together – the observation of the real things that cells do, and their manipulation by genetics (a tool that cell biologists did not have at the time).
And how did you approach the cell biological side of development, particularly since many of the factors involved would have been missed by the screen?
The screen picked switches, decision-making mechanisms: most of these are transcription factors and the cell signalling pathways that govern them. Once you go downstream of decisions, it becomes more complicated and more synthetic, and less easily approachable by a genetic screen that looks for survival, for instance. I think that genetic manipulations are still really powerful here, but you can’t take shortcuts like only looking for genes essential for viability. A number of biological processes go into a change in cell shape, for example, and if you remove one of them, the cell shape transition may not occur in the same way, but the animal may be robust enough that a proportion of embryos survive and you’ll miss it. So understanding morphology turns out to be a process where you use genetics, but need other tools too, and much of this concerns imaging. I think that a major transformation in biology has been the development of microscopes of different kinds, whether confocal or light sheet, or whatever. And digital cameras! To those of us who can remember, images used to be things you had on film and you would spend artistic moments in the darkroom trying to bring out what you wanted; now they are just a matrix of pixels. And I think that aspect – doing microscopy on embryos, and doing it in a quantitative way – meant you learned stuff that you didn’t know before. It’s just transformed the field.
The Wieshcaus lab’s focus on the cell biology of development includes work on nucleolus assembly. In Falahati, et al. (Current Biology, 2016), the dynamics of the nucleolar protein Fibrillarin were tracked in a mutant embryo lacking rDNA repeats
To look at embryos with a quantitative eye, your lab has in recent years incorporated physics and mathematical modelling. From your background, to what extent do you personally need to understand the physics and the maths?
Well I’m totally dependent on having collaborators who have the patience to explain things to me three or four times, and also the tolerance to accept when I decide that I’ve got it, even though there might be a deeper level of understanding available that they feel I haven’t reached yet. But it’s also an interesting question to ask what your thresholds are for understanding: I think you set them, and that sets the style of science that you do. The fact that different scientists have different thresholds is one of the reasons why it is so valuable to collaborate, and also why the social nature of science is so important to its productivity. You profit from talking to people who are interested in working with you, but frankly not willing to invest the same amount of energy into your topic as you do. Of course there’s always danger in not understanding everything, but we live in a dangerous world. Scientifically, if you are going to be on the edge of discovery, you just take the best path forward even if you don’t understand everything in a deeply defendable way.
As someone who has worked on Drosophila for 40 years, what do you think this animal still has to teach us about development?
Science is hard, and in biology we are only at the beginning of a truly deep understanding. To understand development, you work on embryos, and you choose a model organism because to do things well, you need the technologies that everybody else has developed; you need to parasitise a field.
Working with Drosophila today, you have two avenues. If you want to strike out into some totally unexplored area, for instance in something behavioural or evolutionary, flies allow you to do this. And then there are the old areas, things that we think of as established and successful problems, like patterning in the early embryo. What we have there is actually a wonderful cartoon understanding of the process, and this is so rare and valuable in biology because it can provide the basis for building a deeper understanding. So if you want to understand transcription, for example, I think you’d be crazy not to work in flies! In flies you have this organism where you can study a real process with biological meaning that has been selected for in evolution, and which is primed by our existing knowledge for you to start. So the things that we know prime us to go deeper, and for the things that we don’t know, we’re in a position where we can profit from the community and the tools that are available.
In biology we are only at the beginning of a truly deep understanding
It’s now 22 years since you won the Nobel Prize along with Christiane Nüsslein-Volhard and Edward Lewis. What did the award mean to you at the time, and does it still have an impact on your academic life?
Of course it was a wonderful thing to happen for me and my family. It wasn’t really anything I’d foreseen, and then I got ripped out of my normal life for a week in Stockholm. But I also felt like it was wonderful for the community as a whole: at least from my perspective, my colleagues were happy because they saw it as recognition of a set of accomplishments that the field had made.
The other thing that it gave me was a certain power to control my work life. I’ve never assumed major administrative roles at the university – it gave me the power to insist that I would continue to work four hours a day at the bench myself, and meant that people were happy to have me around even if it forced some others to assume extra responsibilities. I could set certain standards for my life: I wanted to be a productive scientist, not to just run a lab and tell other people what to do. I wanted to be the person who did interesting things.
You’ve described yourself as a very visually oriented person, both in terms of science and as someone who loved to draw and paint in your youth. Does this dictate the way you do your science?
Since the public is giving their own money to support science, there are limited resources, an obligation to produce and a certain competition among scientists. So as a young scientist you have to ask yourself – what is my competitive advantage? What am I good at? And almost inevitably that means: what do I enjoy? And the answer will go deeper than science itself. So when I look at most things that I’ve done in the lab, they’ve always had a visual component to them. In the Heidelberg screen, we identified mutants based on our abilities to look at things and recognise what was worth following up on. And to look at gastrulation is a matter of reconstructing and visualising form, and form follows force, so an understanding of physics, of flow and force and mechanics, has, to my mind, at least a strong visual component to it. I am the scientist that I am because of the talents and particular proclivities I have, and clearly painting was another expression of that same quality.
And do you still paint?
I’ve got back in to painting, but it’s challenging because I think to be really good, you have to invest more time than I have. I’m still too hung up on the idea of getting a product at the end, getting the picture to work, rather than doing a hundred versions of it to figure out how to get it to work. Every step in becoming better costs time, which of course is not very different from science, where to be really great, you have to invest and risk, and do things a bunch of times. That’s why science is not a nine to five job: so much of what we do doesn’t quite work out!
What would Development readers be surprised to find out about you?
I am an aggressive card player, and I play to win (though I must say don’t win all the time!). Trudi and I have trained all of our children to be aggressive pinochle players, and also to play to win. Pinochle is different from poker: with poker there is an element of bluff, and while I am a theatrical enough person that I could pull that off, I feel so emotionally dishonest! So I tend towards card games like pinochle where you are confronted with a problem and have to figure out how to win with the cards you have. As long as I don’t have to be dishonest about it!
Check out Development’s collection of print interviews here, and our video interview collection here
A postdoctoral BBSRC funded position is available to study role of Par-3 in E-cadherin recycling and signalling in the group led by Dr Natalia Bulgakova http://www.sheffield.ac.uk/bms/research/bulgakova
The focus of the Bulgakova group is on discovering roles and regulation of cell-cell adhesion using Drosophila as a model organism. We have recently discovered that the protein Par-3 maintains the key cell-cell adhesion protein E-cadherin within the recycling route. The successful applicant will study the mechanism of this Par-3 function and its role in the development and physiology of the animal. The project will employ a combination of molecular genetics, live imaging, and protein biochemistry.
The successful applicant will have a PhD or equivalent experience relevant to studying cell-cell adhesion or intracellular trafficking, knowledge of working with genetic model systems and be highly motivated. Experience in using Drosophila, molecular biology, protein biochemistry, and confocal microscopy, and knowledge of cell-cell adhesion, cell polarity and intracellular trafficking will be an advantage. The successful candidate will be expected to have excellent interpersonal and communication skills, be highly independent, and committed to research in a fast-moving and competitive field.
Fixed-term with an immediate start date and an end date of 31 March 2020
For more information and to apply, please see http://www.jobs.ac.uk/job/BBL246/research-associate/
The publication of Marc Tessier-Lavigne’s seminal Cell papers (1, 2) in 1994 describing the identification of netrin1 (from the Sanskrit word, netr, meaning “one who guides”) was a defining moment in my graduate career. My friends and I talked about those papers for weeks, from the incredible technical feat, the biochemical purification of netrin1 from tens of thousands of chicken brains, to the commonality of the neural developmental mechanism, based on the homology of netrin1 with Unc6, a gene previously identified by Ed Hedgecock and Joe Culotti in a C.elegans screen for axon guidance defects (3).
A couple of years later, I interviewed for postdoctoral positions in San Francisco Bay area in neural development laboratories, and had arranged to stay with Marya Postner, a fellow Princeton graduate alumnus. Marya just happened to be married to Tito Serafini, who, with Tim Kennedy, was one of two lead postdocs on the seminal 1994 Cell papers. Tito was still working with Marc at the time. I asked him breathlessly about the status of the netrin1 project – did they have a netrin1 loss of function phenotype yet? Tito looked happy, triumphant even. “Yes!” he said. “We’ve knocked out the mouse homologue. Commissural axons stall before they reach the floor plate.”
This Cell paper came out a couple of months later (4) and cemented the idea of chemotaxis as the prevailing model of axon guidance (5): netrin1 is secreted from floor plate (FP) cells at the ventral midline of the spinal cord, and like a beacon in a harbor guiding ships in the night, orients commissural axons to grow towards it (Fig. 1A). This model fit with insight dating back as far as Cajal (6), including work performed in grasshoppers (7, 8) and then in vertebrates (9, 10). Together, these studies suggested that axons could be guided in a stepwise manner over long distances by chemotropic attractive or repellent cues diffusing from “guidepost” cells that they encountered along the way. The axon guidance field also recognized that axons also grew on local substrates provided by extracellular matrix components, such as laminin. But their contribution was generally considered prosaic, i.e. passive carpets that permitted or prevented axon growth. The more important and interesting contribution to neural circuit formation came from the chemotropic cues, the netrins, semaphorins, the slit/robo pathway and morphogens.
Problems with the chemotropic model for netrin1
I have taught the chemotropic model of axon guidance for years in my undergraduate and graduate lectures, with netrin1 as the centerpiece discovery. It was a beautiful example of scientific daring being rewarded with mechanistic understanding. I was able to trace a path from the first century-old scientific insight to the netrin1 mutant phenotype that strongly supported that hypothesis.
However after starting my own laboratory in 2004, I began to wonder about a reported, but underappreciated, feature of the netrin1 expression pattern. Textbooks often show the distribution of netrin1 in the chicken spinal cord, where netrin1 is expressed at high levels specifically in the FP. However, my first undergraduates – Joe Herrold and Anna Maglunog – found that mouse netrin1 is also expressed in the ventricular zone (VZ), which is the central compartment where the neural progenitor cells reside, oscillating back and forth on radial process as they proliferate. Spinal axons uniformly avoid growing in the VZ, staying rather at the “sides” of the spinal cord in a region that will ultimately segregate into the grey and white matter. Anna found that netrin1 expression extended into the dorsal VZ and appeared to strengthen, rather than diminish, over time (Fig. 1B).
This distribution had been accurately described in Serafini et al (4). But, it remained unresolved why spinal commissural axons first grew around the domain of VZ-derived netrin1 before growing towards FP-derived netrin1 (Fig. 1A). Were commissural axons unresponsive to ventricular netrin1? In that case, how did commissural axons then become responsive to FP-derived netrin1 to grow towards the ventral midline? Was there a molecular switch that controlled this process? Joe also noticed that in netrin1 mutant mice, while the vast majority of commissural axons of the Tag1+ subtype stalled as they entered the ventral spinal cord as published (4), there were always some Tag1+ axons that entered the VZ. Again, this latter phenotype was reported by Serafini et al, but it was not a major focus of the paper. We also wondered why spinal axons usually grew around the VZ. Was there a repellent in the VZ? Could netrin1 be that repellent?
I wrote some of these ideas into an R01 grant application that was eventually funded in 2008, which allowed me to bring a postdoctoral fellow on board to work on these questions. However, the project stalled for two years; the Tag1+ axon mispolarization phenotype was subtle and it remained stubbornly unclear whether these axons originated from within the spinal cord or from the dorsal root ganglia (DRGs) in the adjacent peripheral nervous system. Moreover, my plan to tackle the problem by recapitulating the putative VZ-repellent activity in a tissue co-culture assay proved challenging. No progress was made and alas, the postdoc left my lab. With time running out to fulfill this aim of my grant, I recruited a student, Supraja (Sup) Varadarajan onto the project with the idea that she could complete the characterization of the netrin1 phenotype. It would be a quick paper, I reassured her. One of the first ideas that we had was to characterize the netrin1 mutant using a wider range of axonal markers.
Figure 1. (A) In the canonical chemotaxis model, axons grow towards a diffusible source of floor plate (FP)-derived netrin1. (B, C) In our haptotaxis model, netrin1 is expressed by neural precursor cells in VZ (red domain) and then netrin1 protein is transported to the pial surface via their radial processes to form a growth substrate (green line). Axons extend adjacent to this substrate in a Dcc-dependent manner. (D) Axon growth is stalled, disoriented, and/or defasciculated in the absence of netrin1 (or Dcc). (E, F) Conditionally ablating netrin1 supports the haptotaxis model: VZ-derived netrin1, not FP-derived netrin1, is required to guide spinal commissural axons. Figure adapted from Varadarajan et al (11).
First moment of clarity: many types of spinal axons invade the VZ in netrin1 mutants
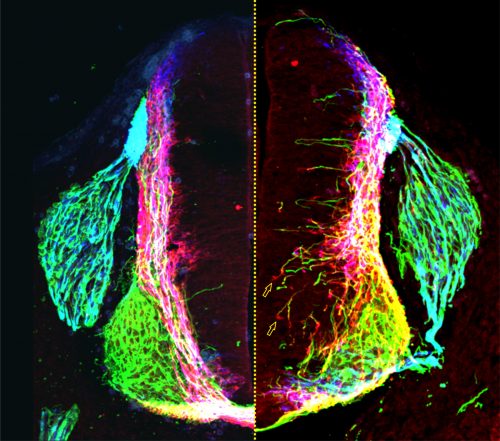
Not long afterwards, Sup called me over to her computer very excitedly: “LOOK at the pattern of neurofilament innervation!” she said. Neurofilament (NF) is an intermediate filament present ubiquitously in axons. Sup had made transverse slices of control and netrin1 mutant spinal cords and stained them with antibodies against NF. While control NF+ axons grew their usual orderly way avoiding the VZ, to our amazement we saw that NF+ axons were now profusely growing into the VZ in the netrin1 mutant (Fig. 1D, Fig. 2). Antibodies against another protein, Robo3, which labels all commissural axons, showed a similar phenotype; axons were no longer tightly bundled or fasciculated. Rather, the axons radiated in all directions, including into the VZ (Fig. 2). Thus, the observed stall of Tag1+ commissural axons appeared to be an anomaly: the loss of netrin1 resulted in other spinal axons extending wildly into the VZ. We suddenly had clear evidence that there might be a repellent in the VZ, with netrin1 as a top candidate for that repellent.
Sup then spent considerable time mapping domains of netrin1 expression in the mouse spinal cord at different stages of development. She found two general patterns of behavior: 1) spinal axons rarely grow on netrin1-expressing cells, and 2) spinal commissural axons grow precisely around the boundary of netrin1-expressing cells in the VZ. But were these activities mediated by a long-range activity from the FP or a more local, short-range activity from the VZ? Our genetic manipulations strongly supported the presence of a VZ-derived netrin1 repellent. First, we characterized Gli2 mutants, which have no FP (12) either singly or in combination with a netrin1 mutation. The result was clear-cut – it was not enough to remove the FP; NF+ axons only invaded the VZ in the absence of VZ-derived netrin1. Second, in collaboration with Jennifer Kong and Bennett (Ben) Novitch, we were able to ablate netrin1 expression from a stripe of neural progenitor cells, thereby creating two de novo netrin1(-):netrin1(+) boundaries. To our amazement, axons now detached from their normal trajectories and extended into the VZ to follow along one of the ectopic boundaries of netrin1 expression. Everyone in the Butler/Novitch joint lab meeting applauded when Sup showed these results for the first time.
Figure 2: Transverse section of a control (left of dotted line) and netrin1 mutant (right of dotted line) mouse embryonic spinal cord, showing NF+ (green), Robo3+ (red) and Tag1+ (blue) axonal staining. Figure adapted from Varadarajan et al (11).
Revised model: netrin1 provides a growth boundary for axon extension
Our first model was that netrin1, present in the VZ, was repulsive for axon growth. But our results were now suggesting a more complex activity. While spinal axons did generally avoid growing on netrin1-expressing cells, commissural axons appeared to grow preferentially along a netrin1(-): netrin1(+) boundary. Since there wasn’t an obvious term for this phenomenon, which was unwieldy to constantly explain, Sup came up with the concept of a “hederal” boundary. Ivy (genus, hedera) uses a wall (c.f. netrin1) as a necessary scaffold for growth, but it is unable to penetrate this wall as it grows. We wondered whether this hederal activity of netrin1 was more attractive or more repulsive, and tested this idea by examining mice lacking different classes of netrin1 receptors, sent to us by Artur Kania.
In the canonical model, netrin1 results in attractive or repulsive responses in axons by activating different receptor complexes. Thus, Dcc translates the attractive responses of netrin1 (13), whereas the Unc5 family mediates the repulsive responses (14). I was confident we would find that a member of the Unc5 family decoded the netrin1 growth boundary in axons, thus confirming that we were describing a repellent activity. But I was wrong: Sup found only minor axon guidance phenotypes in the Unc5 mutants, many of which had been reported before, and stemmed from the loss of Unc5 expression in the DRGs. However, the Dcc mutants looked just like the netrin1 mutants: Tag1 axons stalled, as already described (13) and NF and Robo3 axons dramatically extended into the VZ (Fig. 1D). Thus, Dcc appears to be the chief receptor that mediated the ability of spinal axons to avoid the VZ, and grow alongside a netrin1(-):netrin1(+) border. Moreover, these findings suggested that netrin1-Dcc might be working through an attractive, rather than repulsive, mechanism.
Second moment of clarity: netrin1 protein is deposited on the pial surface of the spinal cord where it acts as a haptotactic growth substrate
As we started to write the paper, Sup had a thesis committee meeting with Kelsey Martin, Alvaro Sagasti and Larry Zipursky where she was questioned skeptically about the feasibility of our hederal model. Larry, in particular, wanted to know more about the distribution of netrin1 protein. Larry, working with his postdoc Orkun Akin, had just shown that netrin had an adhesive, rather than chemotropic, role in the fly medulla (15). We grudgingly admitted that we had never looked, because we had no expectation that netrin1 protein would be anywhere other than the VZ. A couple of days later, Sup appeared at my door, looking worried. “The staining doesn’t look right,” she said. “There’s a lot of background staining on the pial surface of the spinal cord, and in axons.” I internally cursed the non-specificity of our antibody. “Try antigen retrieval.” I said, “That’s what Tim Kennedy had to do!” (16). Sup came back a day or two later, looking even more worried, and told me that the antigen retrieval protocol made the staining “worse.” Both the pial and axonal staining looked brighter than ever. “It looks real!” she said glumly. This distribution made no sense in our model, and I complained to Larry about his opening up a can of worms, when I saw him at a seminar that afternoon. “It’s a bad molecule,” Larry joked.
Sup was right; the antibody staining did look real. There were very low levels of netrin1 in the VZ as we had predicted, but there were much higher levels on most of the pial surface around the circumference of the spinal cord, and on axons (Fig. 1B). I showed James Briscoe the distribution pattern when he visited UCLA a couple of weeks later. “Of course it’s real,” he said, “The neural progenitor cells are making netrin1, and then depositing it on the pial surface using their radial endfeet.” We corralled Ben in from his office next door to mine to assess what he thought. He thought the staining was real too. The three of us peered excitedly at my computer screen, the realization of how the pattern of the netrin1 transcript related to the distribution of netrin1 protein slowly sinking in. We then discussed nothing else for the rest of the weekend.
Sup went on to show that indeed James’ supposition was correct. Through a trick of their cellular geometry, the neural precursors that make netrin1 then use their radial processes to deposit it as a growth substrate onto the pial surface (Fig. 1B, C). No netrin1 is made by the dorsal-most neural precursors, and indeed there is no netrin1 on the most dorsal pial surface. Sup pointed out that the resulting sharp on:off netrin1 boundary on the dorsal pial surface really suggested that netrin1, a member of a laminin family, could not be highly diffusible. In other exhilarating moments of understanding, we realized that spinal neurons specifically initiate NF+ axonal growth on this netrin1 pial-substrate. And that netrin1 only accumulates on commissural axons as they grow adjacent to the netrin1 pial-substrate, as if netrin1 can transfer from this substrate to axons. In a further remarkable result, Sup found that the putative axonal-transfer of netrin1 requires Dcc.
We finally understood how to model our results: VZ-derived netrin1 acts locally as a substrate to promote fasciculated axonal growth in an oriented manner towards the ventral midline. Netrin1 and Dcc appear to cooperate within axons as part of the mechanism that orients growth (Fig. 1C). The innervation of the VZ that we had observed in both netrin1 and Dcc mutants might in fact be randomized, or defasciculated, growth as a result of losing this adhesive interaction (Fig. 1D). VZ-derived netrin1 thus appears to act by haptotaxis, i.e. by the establishment of a local adhesive surface, the alternative model to chemotaxis. As a mechanistic side note: it remains unclear how this adhesive surface acts to orient and fasciculate commissural axon growth and whether there is an additional “no go” activity in the VZ. Is this mechanism functioning solely through haptotaxis? Or is there also a “hederal” component? Nonetheless, these activities – the pull of an adhesive substrate that promotes fasciculation, perhaps coupled with the push of a “no go” signal – permit axons to grow along the netrin1(-):netrin1(+) border, i.e. in a circumferential path precisely around the VZ.
Third moment of clarity: FP-derived netrin1 is dispensable for axon guidance
Our model – the ability of neural progenitor cells to deposit a haptotactic substrate of netrin1, which promotes ventrally-directed, fasciculated axon growth – was a notable departure from the textbook view of netrin1. But was this model the only mode of action? Or did netrin1 have long-range or short-range activities depending on the cell type? In his original papers, Marc Tessier-Lavigne argued the case for and against netrin1 acting by chemotaxis or haptotaxis (1), but ultimately settled on the idea, well supported by the data at the time, that netrin1 was presented as a long-range cue from the FP. I discussed this point vigorously and endlessly with Sup, Ben, Artur, Larry and Orkun. We had not doubted this model until now. Could our phenotypes still be explained by a long-range activity from the FP? Were the activities of short-range VZ-derived netrin1 in addition to the long-range activities? Or could FP-derived netrin1 really be dispensable for axon guidance?
In the end, the reviewers decided it. The beauty of the finding that neural progenitor cells lay down a substrate of netrin1 that orients and promotes the axonal trajectories of their own neural progeny was not considered sufficiently novel. It was clear what experiment needed to be done, thus we requested the conditional netrin1 lines from Holger Eltzschig and set about breeding them to Cre recombinase driver lines that would remove netrin1 specifically from either FP cells (ΔFP, Fig. 1E) or the dorsal VZ (ΔVZ, Fig.1F). Sup accomplished her breeding scheme astonishing quickly and the day came when she finally had the answer sitting on a microscope slide. I ran to the confocal room, both anxious and excited, to discover the result. “There’s no effect,” she said. “It doesn’t matter if you remove netrin1 from the FP!” In contrast, removing netrin1 from the VZ had profound effects. Axons grew in all directions, but only locally, specifically in the region where netrin1 had been removed from the VZ. Together with the Gli2-mediated FP ablation data, our studies had found no evidence for long-range chemotaxis in the spinal cord. Who knew? Perhaps Cajal’s model was wrong!
The paper was finally accepted at Neuron (11), and came out at the same time as a paper from Alain Chedotal in Nature (17), with complementary findings in the hindbrain. A few days later, I ran into a colleague on the street onside my lab. “I saw your paper in Neuron!” she laughingly scolded me, “But please don’t tell me I have to change my lecture on netrin1! I liked that lecture!” I liked my lecture on netrin1 too, but now I also have to change it. Netrin1, of course, remains the supreme architect of spinal circuitry, but now acts locally as a directional surface along which axons can extend, akin to the holds used by climbers to pioneer their way to the top of a mountain.
Samantha Butler
Department of Neurobiology,
Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research,
Intellectual and Developmental Disabilities Research Center
University of California, Los Angeles.
Here are the highlights from the current issue of Development:
A new model for lineage segregation
Lineage segregation during gastrulation has long been thought to be driven by differential cell adhesion and cortical tension among cells, which would together lead to a differential tissue surface tension (TST) and the spatial segregation of specific cell types. However, this long-standing hypothesis is mainly based on in vitro work, and it is as yet unclear whether it holds true in vivo. Now, on p. 1798 Carl-Philipp Heisenberg and colleagues assess the role of differential TST in lineage segregation and find that, contrary to in vitro work, differential TST is insufficient to explain progenitor cell segregation and germ layer formation within the in vivo gastrulating zebrafish embryo. In the study, the authors describe their unique version of video force microscopy called 3D CellFit, which allows them to analyse surface tensions in 3D within a living organism. Using this method, the authors show that ectoderm and mesoderm tissues do not, in fact, exhibit differential TST in the gastrula. They further present evidence that the apparent discrepancy between the in vitro and in vivo results is due to a difference in osmolarity between the culture medium and the interstitial fluid that surround the cells. Finally, by inhibiting the function of the small GTPase Rac, a key regulator of protrusion-driven cell migration, the authors show that directed cell migration, rather than differential TST, provides the major mechanism that determines the segregation of the germ layer progenitors.
Exciting input for inhibitory neurons
A crucial phase in neuronal development is the integration of newborn neurons into circuits. The right balance must be struck between excitatory and inhibitory neurons; however, the mechanisms that control inhibitory neuron integration and drive the maturation of inhibitory connectivity remain largely uncharacterized. In this issue (p. 1807) Michael Francis and colleagues identify a novel, non-cell-autonomous mechanism that regulates inhibitory neuron synapse formation at the neuromuscular junction (NMJ). The authors examine the electrophysiology and structural organization of GABAergic synapses at the NMJ in a number of different C. elegans mutants with developmental or functional defects in excitatory motor neurons. These analyses reveal that the activity of excitatory cholinergic motor neurons, during a period that coincides with the development of postembryonic GABAergic motor neurons, critically affects the size and distribution of GABAergic pre- and post-synaptic specializations. Furthermore, a severe reduction of cholinergic inputs to newly born GABAergic neurons reduces their synaptic density but increases the synapse size. This study makes an important contribution to our understanding of how neuronal activity impacts synapse development and highlights the functional relationship between excitatory and inhibitory neurons during circuit formation.
Heat shock protein regulates human hepatocyte differentiation
The directed differentiation of human induced pluripotent stem cells (iPSCs) into mature hepatocytes is a major goal of liver research. The approach relies on the recapitulation of developmental processes, and thus a better understanding of what regulates hepatocyte differentiation is essential in order to produce these cells more efficiently and to a greater maturity. In this issue (p. 1764) Stephen Duncan and colleagues identify heat shock protein 90 beta (HSP90β) as a novel regulator of endoderm-to-hepatocyte conversion in differentiating human iPSC cultures. The authors begin the study by conducting a screen for small molecules that modify the activity of master hepatocyte transcription factor HNF4A, identifying 132 candidate ‘hits’. They then focus on the role of molecular chaperone HSP90β and show how it acts at the post-translational level to stabilize HNF4A, thus controlling its half-life and availability. Targeted CRISPR-CAS9 mutations in the gene encoding HSP90 perturbs HSP90β levels, resulting in a dramatic reduction of HNF4A protein levels and reduced expression of HNF4A target genes. Moreover, these experiments reveal that HSP90β is specifically required for endoderm-to-hepatocyte conversion, and not for endoderm commitment generally. This study uncovers a new player in hepatocyte differentiation, and further highlights the utility of an iPSC differentiation platform coupled with chemical screens to uncover novel developmental mechanisms.
PLUS
The developmental biology of genetic Notch disorders
This Review discusses the developmental processes underlying Notch-related congenital disorders in humans, drawing on data from model organisms and genome-sequencing projects, on p. 1743.
An interview with Eric Wieschaus
In our latest interview, Eric Wieschaus tells us about his Nobel Prize-winning fly screens, his interest in the cell biology of development and his love of painting, on p. 1740.
Obituary: Tokindo S. Okada (1927-2017)
A retrospective on the life and work of the pioneering Japanese developmental biologist Tokindo Okada, whose research focussed on cell plasticity and transdifferentiation, on p. 1737.
Our lab is interested in the fundamental molecular mechanisms underlying lineage decision-making in stem cells and in vivo. We are fascinated by the question how defined and stable cell types are generated by the interplay of signaling inputs and gene regulatory networks. We study this question by precise quantification of the states of single cells in combination with bioinformatics analysis and machine learning. Based on this quantitative understanding we want to develop new ways to manipulate lineage decisions during in vitro differentiation in precisely controlled ways. Our group is highly interdisciplinary and works at the interface of biology, biophysics, bioinformatics and biomedical sciences.
References:
Semrau, S., van Oudenaarden, A., 2015. Studying Lineage Decision-Making In Vitro: Emerging Concepts and Novel Tools. Annu. Rev. Cell Dev. Biol. 31, 317–345. doi:10.1146/annurev-cellbio-100814-125300
Semrau, S., Goldmann, J., Soumillon, M., Mikkelsen, T.S., Jaenisch, R., van Oudenaarden, A., 2016. Dynamics of lineage commitment revealed by single-cell transcriptomics of differentiating embryonic stem cells. bioRxiv 068288. doi:10.1101/068288
Semrau, S., Crosetto, N., Bienko, M., Boni, M., Bernasconi, P., Chiarle, R., van Oudenaarden, A., 2014. FuseFISH: Robust Detection of Transcribed Gene Fusions in Single Cells. Cell Reports 6, 18–23. doi:10.1016/j.celrep.2013.12.002
Project and key responsibilities
The available postdoc project aims to create a single-cell atlas of the human embryonic kidney. Information about the transcriptional profiles and locations of all cell types in the embryonic kidney will improve our understanding of kidney development and will provide an important benchmark for kidney organoids. In this project you will be responsible for performing single-cell RNA-seq and single-molecule FISH measurements of human embryonic kidney samples. The necessary experimental techniques are established in our lab and samples will be provided by our collaborators. In particular, you will dissociate the tissue and prepare single-cell RNA-seq libraries with the drop-seq technique (Macosko et al., Cell, 2015). You will analyze the RNA-seq data (potentially together with a bioinformatics collaborator) and identify cell types using state-of-the-art machine learning tools. Based on these results you will define a set of marker genes that will allow you to locate cell types by single-molecule FISH in intact tissue sections. This comprehensive spatial molecular data set will then allow you, for example, to establish intercellular signaling networks.
Selection criteria
You hold a PhD degree in one of these disciplines: biology, biochemistry, bioengineering or related disciplines
You have a strong interest in experimental quantitative biology, in particular related to human development and stem cell differentiation
You have experience with molecular biology techniques, in particular NGS library preparation
Experience with programming in R or Matlab and relevant bioinformatics packages is a plus.
You are proficient in spoken and written English, and have good communication and writing skills
You are independent, creative and have team spirit
Research at our department
Our lab is part of the Leiden Institute of Physics (http://www.physics.leidenuniv.nl) and situated at the Leiden Cell Observatory (http://cellobservatory.leidenuniv.nl). The Cell Observatory is a highly collaborative community dedicated to the visualization and understanding of the fundamental molecular mechanisms of life, which is part of the core scientific profile of Leiden University. The Cell Observatory houses state-of-the-art bio-imaging facilities shared among the member labs, which actively develop new methods for the quantitative measurement of single-cell properties.
To apply for this vacancy, please send an email to Dr. Stefan Semrau (semrau@physics.leidenuniv.nl)until June 18. Please include your curriculum vitae, a letter of motivation and the names of 3 potential references.
“It finally got accepted!”, followed by “It’s finally out!” about a month later. I am certain this ‘finally’ feeling about their paper is very familiar to those well-acquainted with the peer review process, and it was no different for our recently published Resource article. The ‘biotagging paper’, as we call it within the Sauka-Spengler lab, is the culmination of several years’ of hard (and often frustrating) work that eventually paid off in more (unexpected) ways than one. Tatjana spearheaded the initial work for biotagging while still at Caltech, by transferring components and approaches she developed in the chicken system into the zebrafish. She worked together with Le and Tatiana, then postdoctoral fellows at Caltech, before the rest of us joined in for the lengthy optimisation, submission and review stage.
Part I: What is “biotagging”?
Biotagging is essentially an encompassing term for our Do-It-Yourself (DIY) in vivo biotinylation system for zebrafish researchers, which can be utilised in creative ways to suit specific biological needs. In vivo biotinylation was first employed in mouse (de Boer et al., 2003) by John Strouboulis when he was in Frank Grosveld’s lab and then applied for use in nuclei isolation from Arabidopsis thaliana by Roger Deal and Steven Henikoff (Deal and Henikoff, 2010). The technique was also applied to the nematode worm at around the same time (Ooi et al. 2009). The core of the technique lies in the ability of bacterial biotin ligase (BirA) to biotinylate an Avi-tagged protein-of-interest. In our binary biotagging system, the researcher decides where BirA will be expressed, which protein is Avi-tagged, and then generates transgenic lines that express these components. Crossing BirA-driver and Avi-effector heterozygous lines will give rise to ~25% of double-alleled offspring, where biotinylation of the Avi-tagged protein product only occurs in cells that also express BirA. The sky is the limit when it comes to the combinations of BirA/Avi that one can use. In the paper, we present a ‘starter’ toolkit consisting of multiple tissue-(neural crest, heart, blood) and cellular compartment-specific (ribosomes, nuclei) transgenic lines, as well as constructs to make your own lines.
Part II: Trials and Tribulations
The deconstruction (and reconstruction) of biotagging
The elegance of in vivo biotinylation means that we are not the only group to perform this method in vertebrates. For example, Michael Housley from Stainier lab (Housley et al. 2014) utilised in vivo biotinylation in zebrafish to apply the TRAP (Translating Ribosome Affinity Purification) method developed by Myriam Heiman and colleagues (Heiman et al., 2008). In vivo biotinylation experiments are not ‘difficult’ per se, but we found that obtaining a clear difference between nuclear and polyribosomal data required a remarkable amount of troubleshooting and optimisation. Our patience paid off, as this was rewarded by a wealth of information provided by a high resolution view into the migratory neural crest nascent (nuclear) and polyribosomal transcriptomes from ~200k cells.
In fact, the entire optimisation process came about by accident. In the paper, we described our surprising results when comparing the nuclear transcriptome of Sox10-positive cells at 16-18ss (migratory neural crest) to a ubiquitous control. By looking at both non-poly and polyadenylated transcripts (whole nuclear transcriptomes), our data did not yield any statistically significant neural crest-specific signature, which is what one would expect, as the enriched transcripts should be neural crest-specific. On the other hand, analysis of polyadenylated nuclear transcripts at 24hpf yielded a neural crest-specific signature. This led to further pain-staking deconstruction of our technique where, months later, we eventually came to the surprisingly simple but crucial element for the protocol to be as consistent as it is today – ensuring the complete lysis of cells (by using hypotonic buffer in excess) to release subcellular compartments into the lysate and minimise the presence of intact cell surface membranes. It is also worth noting, that a key element to the success of our protocol was the usage of an Avi-tagged chicken nuclear envelope protein, RanGAP, to label nuclei. Weirdly enough, chicken RanGAP expressed in zebrafish localised to the nuclei, but zebrafish RanGAP did not.
Having reconstructed the method, we were now eager to repeat the previous 16-18ss neural crest experiment. Imagine our initial dismay when the results were…strikingly similar. However, this was soon replaced by curiosity that drove us to carefully re-examine our results and try to figure out what IS actually going on…
Biotagging of migratory neural crest nuclei transcriptome reveals…what?
The brainstorming sessions were remarkably memorable. They were always long, often ‘lively’ as we picked at each other’s brains, and at times quite outrageous as frustrations ran high. It didn’t take us very long to notice that bidirectional transcription at non-coding regions was enriched in neural crest nuclei. However, it was a long journey after that, as we tried to quantify the phenomenon genome-wide, reproduce what we saw, believe in what we saw, and build our findings into a coherent story. Ultimately, we needed to drive home our main message – that bidirectional transcription at non-coding regions is tissue-specific, thus introducing a new method to detect active regulatory elements. These elements form the molecular signature of neural crest cells, which is traditionally based on the expression of protein-coding genes that are mainly transcription factors. We were also excited to find developmentally regulated long non-coding RNAs and transposable elements.
In short, we are proud of what we have managed to achieve with biotagging. The journey may have been long and arduous, but we have learned a lot from this project. We hope that we have provided a cool new system that includes a fully optimised tool (plasmids on Addgene) with clean protocols (available on the Resources page of our lab website), handy transgenic lines to get started with, as well as analysis pipelines tailored to biotagging datasets. Having worked out the technical intricacies of this system, this toolkit allows the zebrafish community (including us!) to study specific cellular populations in vivo on the systems level, tackling biological questions that could be important to development and disease.
The focus of training will be on construction and automation of image analysis workflows, using as examples more than one toolbox and different exercises. The schools will be held in Gothenburg 11-14th of September 2017, hosted by the Centre for Cellular Imaging – Sahlgrenska Academy, University of Gothenburg, Sweden.
NEUBIAS schools are an excellent opportunity to learn from many experts in bioimage analysis (we are expecting ~40 specialists at the event) and “….a great mix of intensive learning and community networking” (former trainee testimonial!).
applications for Gothenburg are now open (each school has 25 available seats and 10-12 trainers).
Within the COST framework, a few travel grants are offered to applicants who qualify.
Registration deadline: 26th of May, 2017 (must submit also “letter of motivation”).
Selection notification: 1st week of June 2017.
More information about schools (programme & trainers) and venue, travel & lodge available at our website (linked above).
On behalf of all NEUBIAS members, Julien Colombelli, Chair; Kota Miura, Vice-Chair Julia Fernandez-Rodriguez, Local organizer
Carolina Wählby, Jan Eglinger, Joakim Lindblad & Nuno P Martins, TS4&5 programme organizers
Gaby G Martins & Fabrice Cordelières, WG2-Training leaders
NEUBIAS is an European network of currently ~180 members and 35 countries, which aims to promote the communication between Life Scientists, Instrumentalists, Developers and BioImage Analysts and to establish and promote the role of Bioimage Analysts in Life Science. Our mission includes:
A training programme for 3 different target audiences:Early Career researcher, Facility Staff, Analyst (running until 2020 – expected 400 trainees and 15 training schools).
Promote different yearly events (NEUNIAS2020 Conference, workshops [training schools], Taggathons)
Online Resources: Repository of tools and workflows, Benchmarking and Sample datasets, Training material and Open Textbook.
A Short Term Scientific Mission mobility programme for Scientists to visit Host Labs and get in depth insights into cutting edge Image Analysis technology.
Our 20th instalment of this series comes from South Korea and features an investigation into the molecular basis of how temperature influences developmental transitions in Arabidopsis seedlings, recently published in Developmental Cell. We caught up with joint first authors Jun-Ho Ha and Hyo-Jun Lee, and their supervisor Chung-Mo Park, Professor in the Department of Chemistry, Seoul National University (SNU), to hear the story of the paper.
Jun-Ho, Hyo-Jun and Chung-Mo
Chung-Mo, can you give us your scientific biography – I understand you spent some years in the US before returning to South Korea?
CMP I am currently professor in the Department of Chemistry at SNU. I earned my Bachelor of Science in the Department of Science Education from Seoul National University in 1983 and my PhD in molecular virology from State University of New York at Buffalo in 1993 under the supervision of professor Jeremy Bruenn. The topic of my thesis work was identification of killer toxin genes in a double-stranded virus endogenously residing in Ustilago maydis, a corn smut fungus and functional and structural characterization of the killer toxin proteins. After completing my PhD, I worked as a postdoctoral researcher in the same university and the Hauptman-Woodward Medical Research Institute, Buffalo, until I joined the Kumho Life & Environmental Science Laboratory, Korea, as PI in 1996. In the Kumho Laboratory, I worked on the photochemical and photobiological characterization of phytochrome photoreceptors in higher plants and the cyanobacteria Synechocystis PCC6803 and their associated light signal transduction in photomorphogenic responses.
In 2002, I accepted an associate professor position in the Department of Chemistry, SNU, where I have been since that time. While at SNU, my research team has been working on diverse aspects of plant growth and developmental processes, such as seed germination, phase transition and flowering induction, and leaf senescence. I have also been working on plant responses to environmental stresses with emphasis on temperature extremes and drought stresses. In recent years, my research is focused on plant adaptation to high but nonstressful temperatures (warm temperatures) with emphasis on leaf hyponasty, heat dissipation from leaves, and autotrophic development.
And what is South Korea like as a place to do science?
CMP The Korean government and several biotech companies have been investing a huge amount of research fund during the last 30 years. While industrial research and development has been a priority as a potential driving force of economic growth, the Korean government is also spending heavily on basic research. In plant science, there is a national research supporting program, termed New-Generation Biogreen 21, which is organized and supported by the Korean Rural Development Administration. The Program supports various research on both model plants and crops. It is considered that although not sufficient, enthusiastic plant scientists are able to get enough research funds to perform both basic and applied researches in recent years.
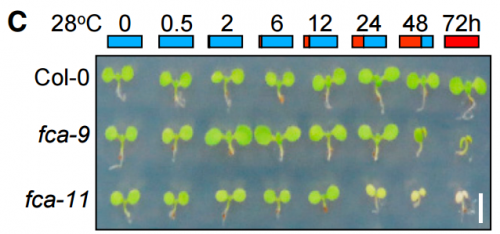
fca mutant seedlings grown at different temperatures, from Figure 1, Ha, et al. 2017
Jun-Ho and Hyo-Jun – how did you come to join Chung-Mo’s lab?
JHH I earned my Bachelor of Science in chemistry. I was also interested in molecular biology with an expectation that combining chemical and biological principles would be exciting in understanding life. While I was looking for an appropriate lab for my graduate study, I met Chung-Mo Park, who is my current thesis advisor. I was greatly impressed by his passion for science and research. It was also impressive that his group is working on plant molecular biology in the Department of Chemistry. I therefore decided to join his laboratory for my graduate study.
HJL Since I was a high school student, I planned to be a scientist with an aim of discovering unknown principles of nature and living organisms. After I entered the Department of Chemistry, Seoul National University, as an undergraduate student, I searched for potential labs in the Department appropriate for my research carrier. I realized that Chung-Mo’ lab is unique among the laboratories in that he is studying plant molecular biology and biochemistry. I thought that understanding molecular biological and biochemical mechanisms underlying plant performance would be helpful for me to find ways to sustain the Earth’s ecosystem. In particular, as a chemist, I thought that applying chemical tools to understanding biological systems would be interesting. I therefore decided to perform my graduate study in his lab.
Before your work, what was known about how plants respond to temperature changes during autotrophic development, and what was the key question you set out to answer?
CMP, JHH & HJL It is well known that extreme temperatures significantly affect plant performance, including autotrophic development. In addition, associated molecular events and signaling schemes are fairly well understood. In nature, the soil temperature is rapidly elevated under warm temperature conditions. Therefore, developing seedlings should cope with high temperatures while they pass through the heat-absorbing soil layer to obtain photosynthetic capacity required for autotrophic growth. However, it is almost unknown how the heat-labile shoot apical meristem tissues of developing seedlings handle the temperature constraints. It has recently been reported that warm temperatures, in a temperature range of 23 – 28oC in Arabidopsis, accelerate cell elongation during early seedling development. Thus, we were curious about whether and how warm temperatures influence chlorophyll biosynthesis during autotrophic development.
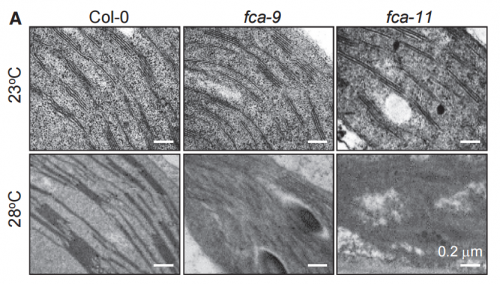
TEM images of cotyledons of 3-day-old seedlings, from Ha, et al. 2017.
Can you give us key results of the paper in a paragraph?
CMP, JHH & HJL We demonstrated that developing seedlings are capable of maintaining chlorophyll biosynthesis required for autotrophic development at warm temperature conditions. A group of photooxidoreductase (POR) enzymes is responsible for chlorophyll biosynthesis. Notably, they are susceptible to warm temperatures and thus rapidly inactivated in developing seedlings while they pass through the warm soil layer. We found that an RNA-binding protein FCA maintains the abundance of POR enzymes at warm temperatures in developing seedlings. Without FCA, plants fail to maintain the enzyme abundance, resulting in loss of chlorophyll and thus failure to achieve autotrophic growth. Our work provide a molecular basis for the acquisition of autotrophic growth under fluctuating temperature conditions in plants.
How do have any idea of what is upstream of FCA? How does it sense temperature changes?
CMP, JHH & HJL Our recent findings strongly support that the typical RNA-binding protein FCA plays a critical role through epigenetic control of target genes during high temperature responses and thermomorphogenesis in Arabidopsis. Our data also indicate that FCA sustains the thermos-stable expression of POR enzymes during autotrophic development at warm temperatures. Altogether, these observations suggest that FCA function is thermos-regulated. However, it is current unclear how FCA is activated by ambient temperatures. We found that gene transcription and protein stability of FCA are not altered by temperature changes. Its subcellular localization is also unaltered under fluctuating temperature conditions.
Our preliminary data suggest that warm temperatures activates FCA through post-translational modifications, such as protein phosphorylation. We are currently under way to examine if FCA is differentially phosphorylated or chemically modified in response to temperature changes by employing global-scale proteomics.
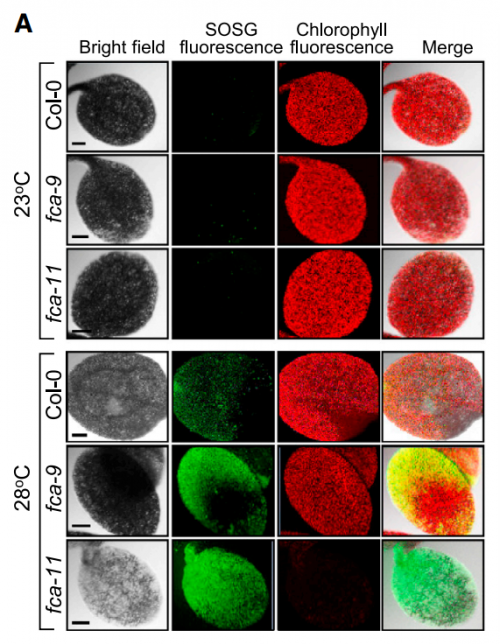
Singlet oxygen accumulation, from Figure 4, Ha, et al. 2017.
Do you think your work will have relevance to agriculture in a warming world?
CMP, JHH & HJL Global warming depicts the gradual elevation of the average temperature of the Earth’s climate system. It is widely documented that under high ambient temperature conditions, plants exhibit distinct morphological and developmental traits, such as accelerated hypocotyl growth, leaf hyponasty, reduction of stomatal density, and early flowering, which profoundly influence crop productivity and commercial values. Our findings on plant thermal responses are closely associated with global warming. We propose that the FCA-mediated thermal adaptation of autotrophic development allows developing seedlings to cope with the heat-absorbing soil surface layer under natural conditions. In particular, we found that a single gene mutation causes a total loss of chlorophyll biosynthesis and autotrophic development at warm temperatures, providing a way of enhancing plant adaptation to thermal fluctuations in crop agriculture.
When doing the research, did you have any particular result or eureka moment that has stuck with you?
HJL & JHH In the initial stage of the research, we germinated and grew the FCA-defective mutants at normal temperatures for 3 days before transferred to warm temperatures to see if the fca mutations affect seedling growth. However, we did not observe any phenotypic differences in seedling growth and greening patterns in the mutants. A few months later, we anticipated that the fca mutations might affect the earlier stages of seedling growth. To examine the hypothesis, we germinated and grew the mutant seedlings at 28oC. We were surprised at the albino phenotype of the mutants. This observation triggered the re-examination of the thermal phenotypes of the fca mutants, resulting in the completion of this paper.
At first, we could not figure out why the fca mutants exhibited albinism only when germinated and grown at warm temperatures. As a potential cause of the albino phenotype, we considered several possibilities, such as defects in chloroplast development, chlorophyll biosynthesis, or both. It was found that the expression of POR genes was disrupted in the fca mutants when grown at warm temperatures. Accordingly, the level of chlorophylls was extremely low in the mutants, showing that the thermo-sensitive albino phenotype of the mutants is caused primarily by defects in chlorophyll biosynthesis, consistent with the FCA-mediated stabilization of POR production.
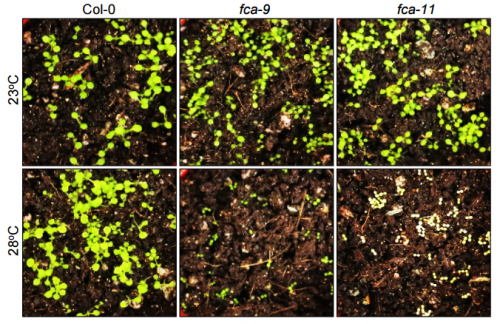
fca seedlings grown in soil, from Figure S6, Ha, et al. 2017
And what about the flipside: any moments of frustration or despair?
HJL & JHH The FCA-defective mutants are well-known late flowering mutants. A set of transgenic fca plants expressing POR genes were required for this study. It needs a lot of time to generate the transgenic plants because it takes 3-4 months to obtain seeds from the transgenic plants. While we were generating transgenic plants, we realized that a wrong expression construct was accidentally used, spending at least 5 additional months to obtain correct transgenic plants.
We also remember the frustrating moment when temperature controllers in the culture room were out of order during last summer, when we experienced a rarely high temperature and thus unstable supply of electricity in Korea. We had to grow a full set of plants again after a period time for fixing the temperature controllers.
What are your career plans following this work?
HJL I am currently a postdoc in Chung-Mo Park’s lab. I will continue studying for a while on molecular and physiological mechanisms underlying plant thermomorphogenesis. I am interested in the as-yet unidentified regulator of POR abundance at warm temperatures. After finishing the experiments, I am planning to find an appropriate postdoc position to extend my research career in environmental control of plant proteomics.
JHH I hope to be able to finish my thesis study in a couple of years, after which I am planning to find postdoc positions in Korea or in USA to extend my research career in the field.
And what next for the Park lab?
CMP We have a well-organized research system with a variety of molecular and biochemical tools, personnel, and facilities. We are specialized in gene regulatory mechanisms with emphasis on induction and activation mechanisms of transcription factors. Using these research tools and system, we will further extend our researches on plant thermomorphogenesis, which is emerging as a hot issue in the field because of the growing concern about global warming. In particular, we are focused on the functional linkage between photomorphogenic responses and growth hormones. We are also preparing a long-term project for engineering crop plants to enhance their adaptation capacity to changing temperature environment.

 (1 votes)
(1 votes)




 (No Ratings Yet)
(No Ratings Yet)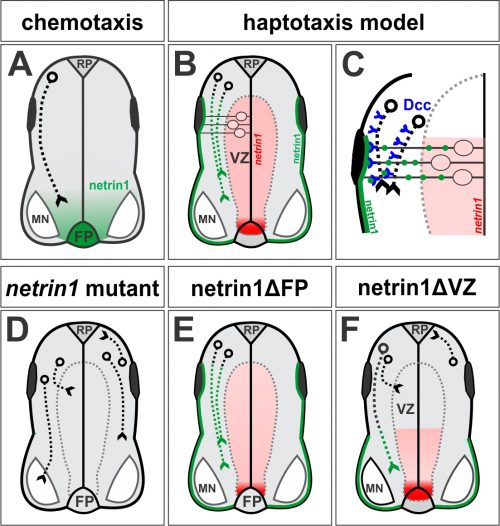
 (16 votes)
(16 votes)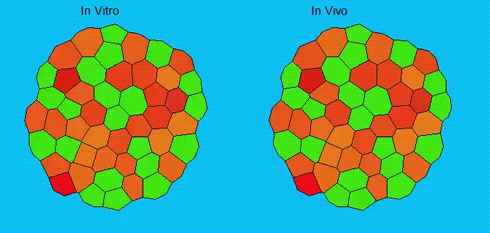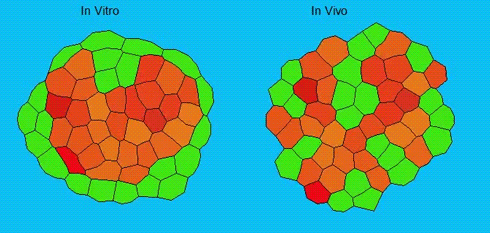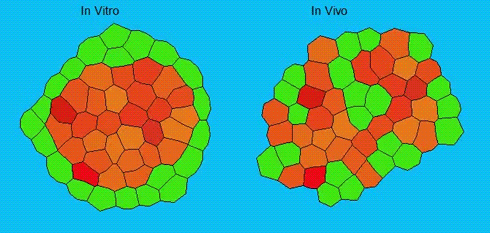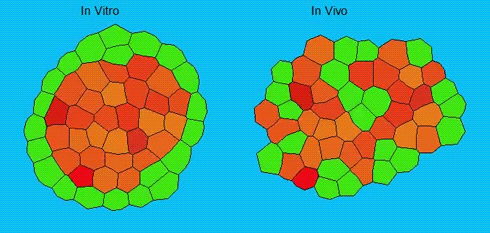
 In this issue (p.
In this issue (p.  In this issue (p.
In this issue (p. 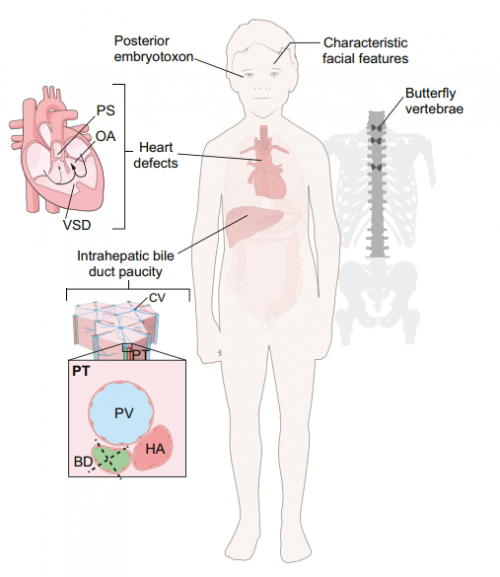


 NEUBIAS, the Network of European
NEUBIAS, the Network of European