This interview by Aidan Maartens first featured in Development, Volume 143, Issue 22.
Kathryn Anderson is Professor and Chair of the Developmental Biology Program at the Sloan Kettering Institute in New York. Her lab investigates the genetic networks underlying the patterning and morphogenesis of the early mouse embryo. We caught up with Kathryn at the 2016 Society for Developmental Biology – International Society of Differentiation joint meeting in Boston, where she was awarded the Edwin G. Conklin medal.

You’re here at the SDB-ISD meeting to receive the Edwin G. Conklin medal. What does the award mean to you?
The recognition of your colleagues is a wonderful thing and I guess I’m kind of sentimental about this. It’s nice to think that the people in the field appreciate what we’ve done. The other part that’s particularly nice is that for a long time I was thought of as a fly person who’d gone astray! Doing the mouse project was a big risk and to think that people have appreciated it means a lot to me.
When did you first become interested in biology?
My parents must have steered me in this direction. My father was probably a frustrated chemist and I guess he wanted me to do science, not that I was particularly aware of it. I did science fairs at junior high and then I had an absolutely wonderful biology teacher in high school: he made a huge difference. We’d read things like The Double Helix and got an impression of what it was like to do science, not just the theory. And also, I grew up by the beach so a lot of my spare time was spent looking at tide pools, pulling up little animals – I always loved that.
In your early career at Berkeley you made seminal contributions to our understanding of early embryonic patterning in Drosophila. Looking back, do you think many of the questions you grappled with then have now been answered?
At the time I moved away from dorsoventral patterning, there were many open questions in the field, but I think no burning ones that I particularly wanted to get to the bottom of. Since I’ve left the field there have been huge steps forward. I’ve particularly followed progress in understanding morphogenesis, for instance Maria Leptin’s and Eric Wieschaus’ work on how the furrow cells actually do what they need to do, which has been really exciting. The Toll pathway has also opened up in exciting new ways. So it’s great to be able to follow the field in depth, because I used to live there!
In the 1990s your lab switched from working exclusively with flies to accommodate mice as well and mice are now your lab’s primary model system. What spurred on that change, and was it a challenging transition?
I was always interested in mammalian development and I took a mouse genetics course at Bar Harbor when I was teaching in Berkeley. So I was intrigued by the possibilities of mouse genetics. And then I did this totally wonderful sabbatical with Rosa Beddington at Mill Hill in London. At the time Mill Hill was exclusively vertebrate developmental biology, so to go there from the invertebrate field was very engaging. Rosa was amazing and she made mouse embryos seem totally accessible. I had some time to think on this sabbatical and thought ‘well, these guys really know what they’re doing, I can’t really improve on mouse knockout technology’. So what could I do in mice that would be a little different? This was the time when the MIT mouse genome project was beginning, where they’d made DNA polymorphism-based markers for genetic mapping. I read those papers and thought: you could actually map a mutant with a very small number of animals with that technology. So this opened up the possibility of doing forward genetics in mouse. One graduate student, Andrew Kasarskis, was convinced that he could get a thesis out of it, though many people thought I’d lost my mind! He moved with me when we moved the lab to New York, which really helped.
In terms of the challenges: I wasn’t really put off by them. The long generation time didn’t bother me – I mean a fly screen takes a year and a half! You just do things on a smaller scale. My goal was never to do Drosophilasaturation mutagenesis, that wouldn’t have worked.
And you seem to have brought an enthusiasm for genetics into your mouse work…
Genetics is essential. But we’ve never done only genetics – we combined it with biochemistry, cell biology, molecular biology. Genetics doesn’t exist in isolation, but without genetics you don’t know where your reality is. The fun and kind of sad thing about mammalian genetics is that there’s so much we don’t know, so you can do a simple, forward genetic recessive loss-of-function screen and still find new stuff. And this is in the mouse, which is such an important model – it’s a real thrill.
And what got you interested in primary cilia?
It was totally driven by the screen we did. It turns out there are a lot of genes in the mammalian genome that are required to make primary cilia. And if you make mutations in these genes you get aberrant Hedgehog signalling, and Hedgehog mutants have these beautiful and striking phenotypes at mid-gestation, the stage we chose to screen at. The phenotypes just leap out at you. We were also fortunate that we were prepared to recognise they were Hedgehog signalling mutants. Jonathan Eggenschwiler, the first postdoc to work on mice in my lab, had worked on one of the very first five mutants that came from the screen, which turned out to be Rab23. Jonathan did a lot of painstaking work to show that this mutant disrupted Hedgehog signalling and in the course of doing that we got to know what Hedgehog pathway mutants looked like morphologically.
Other people had made IFT [intraflagellar transport] mutants that disrupted cilia and hadn’t recognised what they were, but when we saw them in our labs, under our microscopes we said, ‘that’s Hedgehog’! That turned out to be pretty amazing, actually: there’s this whole organelle required for Hedgehog signalling in vertebrates, but not in flies, and there are literally hundreds of non-redundant genes in the mammalian genome where mutations disrupt cilia and thus Hedgehog signalling. It’s a geneticist’s dream, but raises the question of why organise the genome like this: there are so many weak points in Hedgehog signalling – and Hedgehog is so vital.
So at first we had an embarrassment of riches, trying to figure out how each of these genes worked. Other people started working on it too and showed that the Hedgehog pathway proteins are enriched in cilia and that they traffic from one place to another in response to Hedgehog ligands. This became an organelle that’s doing something dynamic and extremely interesting. And then, if you perturb the ciliary structure in different ways, you alter the pathway in different ways. So you can get gain or loss of signalling by disrupting different proteins, all of which are required to make cilia. It became a complex, but also tractable, puzzle.
Your lab also currently works on the mechanisms of gastrulation in the early mouse embryo.
Gastrulation was really what I came into the mouse system to do. To my great disappointment the Toll pathway does nothing in the early mouse embryo: the plan for my sabbatical was to start getting a foothold in what Toll does in early mammalian development, but my and other people’s studies made it pretty clear early on that the conserved function of the Toll pathway was in immunity, not development.
And so we worked on immunity for a little while, but I’m really a developmental biologist. My rationale for doing mouse genetics was that if the Toll pathway has nothing to do with mammalian development, then maybe there are things about early mammalian development that you can’t figure out by extrapolating from flies. So I wanted to find out the rules that control gastrulation and cell fate specification in the mouse. Mice are obviously very different to flies: everything is dynamic. It’s not like a blastoderm where every cell sort of knows what it’s going to do. Cell fate decisions are being made ‘on the fly’: cells move from one place to another, see another cue, do something different. It seemed like the rules might be quite different.
In the meantime a lot of people knocked out a lot of genes and found a bunch of signalling pathways important for mouse gastrulation, but the mutations we identified through screens were mostly other things: cell biology was really prominent. For instance, we’ve been very interested in how epithelial organisation and cell migration are regulated by those signalling molecules, and how they actually control a cell’s behaviour to get it to the right place to sense the next signal. Unlike the famous fly screens, in mouse screens we actually get a lot of the ‘mechanical’-type factors like cytoskeletal regulators. This is partly due to the lack of a real maternal contribution: you’re looking at the first time a lot of these systems are used. One of the first mutants we identified was a regulator of the Arp2/3 complex, something you’d not expect to find as a regulator of patterning in the fly screens, but which had anterior-posterior duplications in the body plan in the mice. This tells us that cell migration is crucial to make the body plan of the mouse. It was surprising and delightful to get these sorts of core cell biological factors coming from the screens.
Your lab is based at the Sloan Kettering Institute. I wonder what working in proximity to cancer biologists is like?
The Sloan Kettering Institute is the basic science branch of the Memorial Sloan Kettering Cancer Center and while there is cancer biology done here, most of it is basic science. When I arrived, I went to the Molecular Biology programme which was this funny mix of nucleic acid biochemists and fly geneticists. When Harold Varmus became president he decided that because of his personal experience, developmental biology was really important for cancer and so there should be a developmental biology unit. I think development is well appreciated in the Institute and we do get exposed to cancer biology. It’s nice to be able to see the connections and learn to speak the language.
Two years ago you were awarded FASEB’s Excellence in Science Award, which recognises outstanding achievements by women in biology. What do you think of the current outlook for young female researchers in developmental biology?
Things have improved, though perhaps not as much or as quickly as one would have hoped. Women face the same challenges as they did 20 years ago and there are fewer women in the field than there should be. I think things get better when there are women in leadership positions and that’s one way forward – to break these glass ceilings and make sure that we lobby to have women in charge, or create positive reinforcements for institutions that take these actions. As well as being positive role models, women in leadership positions can practically understand how to increase the prospects for younger women researchers.
Do you have any advice for young scientists?
Find what you love – it might be something that you love just for a personal reason – and follow that. You really need a personal love for what you’re doing.
What might Development readers be surprised to find out about you?
Maybe it’s not ‘surprising’, but a real passion I have outside the lab is music, whether modern classical composers like David Lang or bands like Radiohead. And New York is a great place for this passion.
 (3 votes)
(3 votes)
 Loading...
Loading...
 (No Ratings Yet)
(No Ratings Yet)
 embryonic microcephaly and has been declared a global health emergency by the World Health Organization. Disruption of the neural progenitor cells is considered to be the major cause of microcephaly; however, the fate of other cell types, including differentiated neurons and vascular cells, remains unknown. Since infected mouse embryos die perinatally, it also remains unknown whether ZIKV infection can cause postnatal microcephaly in animal models. Now, on p.
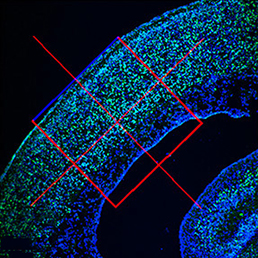
embryonic microcephaly and has been declared a global health emergency by the World Health Organization. Disruption of the neural progenitor cells is considered to be the major cause of microcephaly; however, the fate of other cell types, including differentiated neurons and vascular cells, remains unknown. Since infected mouse embryos die perinatally, it also remains unknown whether ZIKV infection can cause postnatal microcephaly in animal models. Now, on p.  Initiation and subsequent growth of the mammalian tooth depends on distinct populations of epithelial and mesenchymal stem cells located in the labial cervical loop (LaCL) and the neurovascular bundle, respectively. In rodents, Sox2 marks the dental epithelial stem cells (DESCs) and has been shown to be an important regulator of tooth development, but the molecular mechanism by which this occurs has not been determined. In this issue (p.
Initiation and subsequent growth of the mammalian tooth depends on distinct populations of epithelial and mesenchymal stem cells located in the labial cervical loop (LaCL) and the neurovascular bundle, respectively. In rodents, Sox2 marks the dental epithelial stem cells (DESCs) and has been shown to be an important regulator of tooth development, but the molecular mechanism by which this occurs has not been determined. In this issue (p.  Kathryn Anderson is Professor and Chair of the Developmental Biology Program at the Sloan Kettering Institute in New York. Her lab investigates the genetic networks underlying the patterning and morphogenesis of the early mouse embryo. We caught up with Kathryn at the 2016 Society for Developmental Biology – International Society of Differentiation joint meeting in Boston, where she was awarded the Edwin G. Conklin medal.
Kathryn Anderson is Professor and Chair of the Developmental Biology Program at the Sloan Kettering Institute in New York. Her lab investigates the genetic networks underlying the patterning and morphogenesis of the early mouse embryo. We caught up with Kathryn at the 2016 Society for Developmental Biology – International Society of Differentiation joint meeting in Boston, where she was awarded the Edwin G. Conklin medal.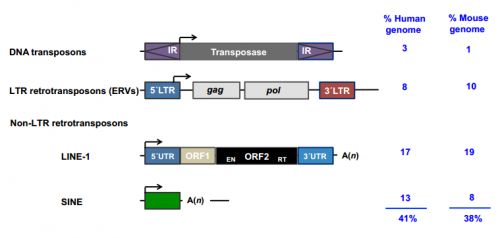 This Review discusses how and when transposable elements are expressed during development and how they modulate genome architecture, gene regulatory networks and protein function during embryogenesis.
This Review discusses how and when transposable elements are expressed during development and how they modulate genome architecture, gene regulatory networks and protein function during embryogenesis.

