February in preprints
Posted by the Node, on 2 March 2020
Welcome to our monthly trawl for developmental biology (and related) preprints.
February’s haul features blooming orchids, regenerating lungfish and human intestines, and also lots of interesting stuff in the ‘Research practice and education section’, from preprint impacts to the dominance of the English language in science.
They were hosted on bioRxiv and arXiv. Let us know if we missed anything. Use these links to get to the section you want:
Developmental biology
| Stem cells, regeneration & disease modelling
Evo-devo & evo
Cell biology
Modelling
Tools & resources
Research practice & education
Why not…
Developmental biology
| Patterning & signalling
Insulin-like signalling influences the coordination of Drosophila larval hemocyte number with body size
Daniel Bakopoulos, Lauren Forbes Beadle, Katherine M. Esposito, Christen K. Mirth, Coral G. Warr, Travis K. Johnson
Mitochondria regulate Drosophila intestinal stem cell differentiation through FOXO
Fan Zhang, Mehdi Pirooznia, Hong Xu
Ecdysone steroid hormone remote controls intestinal stem cell fate decisions via the PPARγ-homologue E75B in Drosophila
Lisa Zipper, Denise Jassmann, Bastian Görlich, Tobias Reiff
Hedgehog Signalling Modulates Glial Proteostasis and Lifespan
Andrew Rallis, Juan A. Navarro, Mathias Rass, Amélie Hu, Serge Birman, Stephan Schneuwly, Pascal P. Thérond
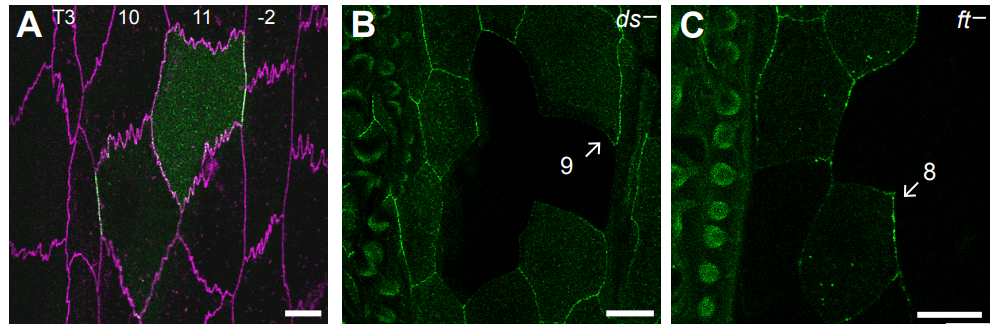
How do Dachsous and Fat polarise cells in the larval abdomen of Drosophila?
Stefano Pietra, KangBo Ng, Peter A Lawrence, Jose Casal
Escargot is involved in labial and antennal imaginal disc development through two different developmental pathways.
Fernando Rosales, Ivan Sanchez-Diaz, Enrique Reynaud, Veronica Narvaez
Dpp and Hedgehog promote the Glial response to neuronal damage in the developing Drosophila Visual system
Sergio B. Velarde, Alvaro Quevedo, Carlos Estella, Antonio Baonza
The Drosophila FUS ortholog cabeza promotes adult founder myoblast selection by Xrp1-dependent regulation of FGF signaling
Marica Catinozzi, Moushami Mallik, Marie Frickenhaus, Marije Been, Céline Sijlmans, Divita Kulshrestha, Ioannis Alexopoulos, Manuela Weitkunat, Frank Schnorrer, Erik Storkebaum
A novel temporal identity window generates alternating cardinal motor neuron subtypes in a single progenitor lineage
Austin Seroka, Rita M Yazejian, Sen-Lin Lai, Chris Q Doe
Continual inactivation of genes involved in stem cell functional identity stabilizes progenitor commitment
Noemi Rives-Quinto, Hideyuki Komori, Derek H. Janssens, Shu Kondo, Qi Dai, Adrian W. Moore, Cheng-Yu Lee
Odd-skipped controls neurite morphology and affect cell survival in Drosophila Melanogaster CNS
Yeoh Sue Lynn, Alina Letzel, Clemence Bernard Hannah Somerfield, Kyle Kyser, Emily Lin, Amanda Roper, Yucen Yuan, Chloe Saunders, Mina Farag, Samual Colourous, Camilla W. Larsen
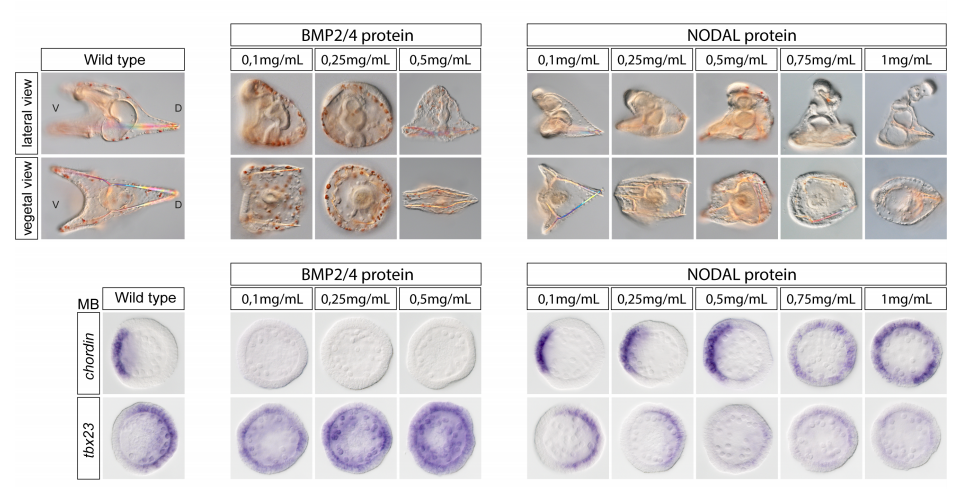
Deciphering and modelling the TGF-β signalling interplays specifying the dorsal-ventral axis of the sea urchin embryo
Swann Floc’hlay, Maria Dolores Molina, Céline Hernandez, Emmanuel Haillot, Morgane Thomas-Chollier, Thierry Lepage, Denis Thieffry
Growth factor-mediated coupling between lineage size and cell fate choice underlies robustness of mammalian development
Néstor Saiz, Laura Mora-Bitria, Shahadat Rahman, Hannah George, Jeremy P Herder, Jordi García-Ojalvo, Anna-Katerina Hadjantonakis
Loss of floor plate Netrin-1 impairs midline crossing of corticospinal axons and leads to mirror movements
Oriane Pourchet, Marie-Pierre Morel, Quentin Welniarz, Nadège Sarrazin, Fabio Marti, Nicolas Heck, Cécile Galléa, Mohamed Doulazmi, Sergi Roig Puiggros, Juan Antonio Moreno-Bravo, Marie Vidailhet, Alain Trembleau, Philippe Faure, Alain Chédotal, Emmanuel Roze, Isabelle Dusart
Control of spontaneous activity patterns by inhibitory signaling in the developing visual cortex
Alexandra H. Leighton, Gerrit J. Houwen, Juliette E. Cheyne, Paloma P. Maldonado, Fred De Winter, Christian Lohmann
Early neurulation recapitulated in assemblies of embryonic and extraembryonic cells
Noémie M. L. P. Bérenger-Currias, Maria Mircea, Esmée Adegeest, Patrick R. van den Berg, Marleen Feliksik, Mazène Hochane, Timon Idema, Sander J. Tans, Stefan Semrau
Moonlighting α-PheRS connects JAK/STAT with Notch signaling for intestinal homeostasis
Manh Tin Ho, Jiongming Lu, Beat Suter
Notch Signaling Commits Mesoderm to the Cardiac Lineage
Evan S. Bardot, Bharati Jadhav, Nadeera Wickramasinghe, Amélie Rezza, Michael Rendl, Andrew J. Sharp, Nicole C. Dubois
Activation of WNT signaling restores the facial deficits in a zebrafish with defects in cholesterol metabolism
Victoria L. Castro, Nayeli G. Reyes-Nava, Brianna B. Sanchez, Cesar G. Gonzalez, Anita M. Quintana
Zebrafish Kit ligands cooperate with erythropoietin to promote erythroid cell expansion
Jana Oltova, Ondrej Svoboda, Olga Machonova, Petra Svatonova, David Traver, Michal Kolar, Petr Bartunek
Bone morphogenetic protein signaling regulates Id1 mediated neural stem cell quiescence in the adult zebrafish brain via a phylogenetically conserved enhancer module
Gaoqun Zhang, Marco Ferg, Luisa Lubke, Masanari Takamiya, Tanja Beil, Victor Gourain, Nicolas Diotel, Uwe Strahle, Sepand Rastegar
The Hedgehog Co-Receptor BOC Differentially Regulates SHH Signaling During Craniofacial Development
Martha L. Echevarría-Andino, Benjamin L. Allen
Gain-of-function mutation in Gli3 causes ventricular septal defects
Antonia Wiegering, Paniz Adibi, Ulrich Rüther, Christoph Gerhardt
LSD1 represses a neonatal/reparative gene program in adult intestinal epithelium
Rosalie T. Zwiggelaar, Håvard T. Lindholm, Madeleine Fosslie, Marianne T. Pedersen, Yuki Ohta, Alberto Díez-Sánchez, Mara Martín-Alonso, Jenny Ostrop, Mami Matano, Naveen Parmar, Emilie Kvaløy, Roos R. Spanjers, Kamran Nazmi, Morten Rye, Finn Drabløs, Cheryl Arrowsmith, John Arne Dahl, Kim B. Jensen, Toshiro Sato, Menno J. Oudhoff
Foxg1 Organizes Cephalic Ectoderm to Repress Mandibular Fate, Regulate Apoptosis, Generate Choanae, Elaborate the Auxiliary Eye and Pattern the Upper Jaw
Claudia Compagnucci, Michael J. Depew
FGF2 modulates simultaneously the mode, the rate of division and the growth fraction in cultures of Radial Glia
Mario Ledesma-Terrón, Nuria Peralta-Cañadas, David G. Míguez
Fgf4 is critical for maintaining Hes7 levels and Notch oscillations in the somite segmentation clock
Mark Lewandoski, Matthew J Anderson, Valentin Magidson, Ryoichiro Kageyama
Small-molecule inhibition of Lats kinases promotes Yap-dependent proliferation in postmitotic mammalian tissues
Nathaniel Kastan, Ksenia Gnedeva, Theresa Alisch, Aleksandra Petelski, David Huggins, Jeanne Chiaravalli, Alla Aharanov, Avraham Shakked, Eldad Tzahor, Aaron Nagiel, Neil Segil, Albert J. Hudspeth
Yap haploinsufficiency leads to Müller cell dysfunction and late-onset cone dystrophy
Christel Masson, Diana Garcia-Garcia, Juliette Bitard, Elodie Grellier, Jerome E Roger, Muriel Perron
β-catenin-Mediated Wnt Signal Transduction Proceeds Through an Endocytosis-Independent Mechanism
Ellen Youngsoo Rim, Leigh Katherine Kinney, Roel Nusse
Quantitative analyses reveal extracellular dynamics of Wnt ligands in Xenopus embryos
Yusuke Mii, Kenichi Nakazato, Chan-Gi Pack, Yasushi Sako, Atsushi Mochizuki, Shinji Takada, Masanori Taira
Defective heart chamber growth and myofibrillogenesis after knockout of adprhl1 gene function by targeted disruption of the ancestral catalytic active site
Stuart J. Smith, Norma Towers, Kim Demetriou, Timothy J. Mohun
Lgr5+ telocytes are a signaling hub at the intestinal villus tip
Keren Bahar Halpern, Hassan Massalha, Rachel K. Zwick, Andreas E. Moor, David Castillo-Azofeifa, Milena Rozenberg, Lydia Farack, Adi Egozi, Dan R. Miller, Inna Averbukh, Yotam Harnik, Noa Weinberg-Corem, Frederic J. de Sauvage, Ido Amit, Ophir D. Klein, Michal Shoshkes-Carmel, Shalev Itzkovitz
Vascular-Derived SPARC and SerpinE1 Regulate Interneuron Tangential Migration and Accelerate Functional Maturation of Human Stem Cell-Derived Interneurons
Matthieu Genestine, Daisy Ambriz, Gregg W. Crabtree, Anna Molotkova, Michael Quintero, Angeliki Mela, Saptarshi Biswas, Peter Canoll, Gunnar Hargus, Dritan Agalliu, Joseph A. Gogos, Edmund Au
Glial type specific regulation of CNS angiogenesis by HIFα-activated different signaling pathways
Sheng Zhang, Bokyung Kim, Xiaoqing Zhu, Xuehong Gui, Yan Wang, Zhaohui Lan, Preeti Prabhu, Kenneth Fond, Aijun Wang, Fuzheng Guo
A gatekeeping role of ESR2 to maintain the primordial follicle reserve
V. Praveen Chakravarthi, Subhra Ghosh, Katherine F. Roby, Michael W. Wolfe, M. A. Karim Rumi
The influence of maternal malnutrition on folate and inositol production and transport in the placenta and gut – a mechanism for fetal growth restriction and fetal disorders?
Elia Palladino, Tim Van Mieghem, Kristin L. Connor
Clustered γ-Protocadherins Regulate Cortical Interneuron Programmed Cell Death.
Walter R Mancia Leon, Julien Spatazza, Benjamin Rakela, Ankita Chatterjee, Viraj Pande, Tom Maniatis, Andrea R Hasenstaub, Michael P Stryker, Arturo Alvarez-Buylla
Urethral luminal epithelia are castration-insensitive progenitors of the proximal prostate
Diya B Joseph, Gervaise H Henry, Alicia Malewska, Nida Iqbal, Hannah M Ruetten, Anne E Turco, Lisa L Abler, Simran K Sandhu, Mark T Cadena, Venkat S Malladi, Jeffrey C Reese, Ryan J Mauck, Jeffrey C Gahan, Ryan C Hutchinson, Claus G Roehrborn, Linda A Baker, Chad M Vezina, Douglas W Strand
“Enteric glia as a source of neural progenitors in adult zebrafish”
Sarah McCallum, Yuuki Obata, Evangelia Fourli, Stefan Boeing, Christopher J Peddie, Qiling Xu, Stuart Horswell, Robert Kelsh, Lucy Collinson, David Wilkinson, Carmen Pin, Vassilis Pachnis, Tiffany Heanue
Zebrafish Kit ligands cooperate with erythropoietin to promote erythroid cell expansion
Jana Oltová, Ondřej Svoboda, Olga Machoňová, Petra Svatoňová, Michal Kolář, Petr Bartůněk
| Morphogenesis & mechanics
Cadherin clustering controls heterogeneous, asymmetric junction dynamics during vertebrate axis elongation
Robert Huebner, Abdul N Malmi-Kakkada, Sena Sarikaya, Shinuo Weng, Dave Thirumalai, John B. Wallingford
An asymmetry in the frequency and position of mitosis in the epiblast precedes gastrulation and suggests a role for mitotic rounding in cell delamination during primitive streak epithelial-mesenchymal transition
Evangéline Despin-Guitard, Navrita Mathiah, Matthew Stower, Wallis Nahaboo, Elif Sema Eski, Sumeet Pal Singh, Shankar Srinivas, Isabelle Migeotte
Coordinated assembly and release of adhesions builds apical junctional belts during de novo polarisation of an epithelial tube
Andrew Symonds, Clare Buckley, Charlotte Williams, Jon Clarke
Buckling of epithelium growing under spherical confinement
Anastasiya Trushko, Ilaria Di Meglio, Aziza Merzouki, Carles Blanch-Mercader, Shada Abuhattum, Jochen Guck, Kevin Alessandri, Pierre Nassoy, Karsten Kruse, Bastien Chopard, Aurélien Roux
Keratins and Plakin family cytolinker proteins control the length of epithelial microridge protrusions
Yasuko Inaba, Vasudha Chauhan, Aaron Paul van Loon, Lamia Saiyara Choudhury, Alvaro Sagasti
Mechanics regulate human embryonic stem cell self-organization to specify mesoderm
Jonathon M. Muncie, Nadia M.E. Ayad, Johnathon N. Lakins, Valerie M. Weaver
Hyaluronan-NK cell Interaction Controls the Primary Vascular Barrier during Early Pregnancy
Ron Hadas, Eran Gershon, Aviad Cohen, Sima Stroganov, Ofir Atrakchi, Shlomi Lazar, Ofra Golani, Bareket Dassa, Michal Elbaz, Gadi Cohen, Elena Kartvelishvily, Raya Eilam, Nava Dekel, Michal Neeman
Par3A and Par3B orchestrate podocyte architecture by regulating RhoA levels
Sybille Koehler, Johanna Odenthal, David Unnersjö Jess, Martin Höhne, Christian Jüngst, Ferdi Grawe, Martin Helmstädter, H. Henning Hagmann, Gerd Walz, Wilhelm Bloch, Carien Niessen, Bernhard Schermer, Andreas Wodarz, Barry Denholm, Thomas Benzing, Sandra Iden, Paul Thomas Brinkkoetter
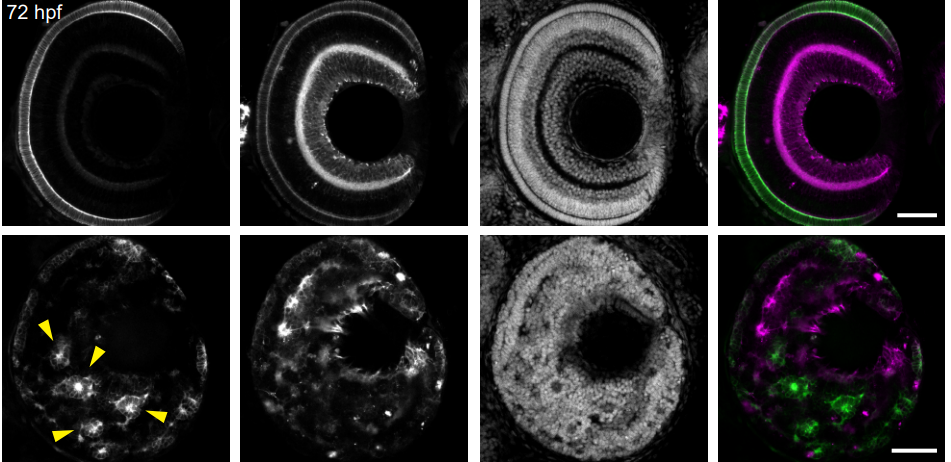
Photoreceptor progenitor dynamics in the zebrafish embryo retina and its modulation by primary cilia and N-cadherin
Gonzalo Aparicio, Magela Rodao, José L. Badano, Flavio R. Zolessi
Transcriptional control of apical protein clustering drives de novo cell polarity establishment in the early mouse embryo
Meng Zhu, Peizhe Wang, Charlotte E. Handford, Jie Na, Magdalena Zernicka-Goetz
TRIM67 Regulates Exocytic Mode and Neuronal Morphogenesis via SNAP47
Fabio L. Urbina, Shalini Menon, Dennis Goldfarb, M. Ben Major, Patrick Brennwald, Stephanie L. Gupton
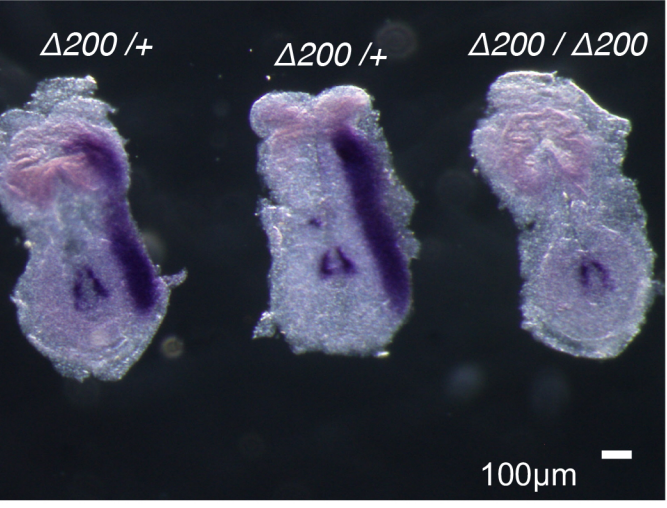
Fluid flow-induced left-right asymmetric decay of Dand5 mRNA in the mouse embryo requires Bicc1-Ccr4 RNA degradation complex
Katsura Minegishi, Benjamin Rothé, Kaoru R. Komatsu, Hiroki Ono, Yayoi Ikawa, Hiromi Nishimura, Emi Miyashita, Katsuyoshi Takaoka, Kana Bando, Hiroshi Kiyonari, Tadashi Yamamoto, Hirohide Saito, Daniel B. Constam, Hiroshi Hamada
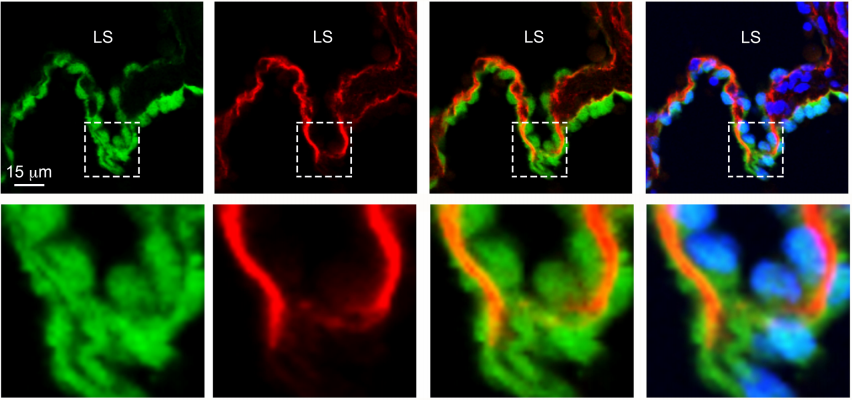
RASA1-driven cellular export of collagen IV is required for the development of lymphovenous and venous valves in mice
Di Chen, Xin Geng, Philip E. Lapinski, Michael J. Davis, R. Sathish Srinivasan, Philip D. King
Prmt5 promotes vascular morphogenesis in zebrafish independently of its methyltransferase activity
Aurelie Quillien, Manon Boulet, Severine Ethuin, Laurence Vandel
S1PR1 regulates the quiescence of lymphatic vessels by inhibiting laminar shear stress-dependent VEGF-C signaling
Xin Geng, Keisuke Yanagida, Rachel Akwii, Dongwon Choi, Lijuan Chen, YenChun Ho, Boksik Cha, Md Riaj Mahamud, Karen Berman de Ruiz, Hirotake Ichise, Hong Chen, Joshua Wythe, Young-Kwon Hong, Constantinos Mikelis, Timothy Hla, R. Sathish Srinivasan
NRP2 as an emerging angiogenic player; promoting endothelial cell adhesion and migration by regulating recycling of α5 integrin
Abdullah AA Alghamdi, Christopher J Benwell, Samuel J Atkinson, Jordi Lambert, Stephen D Robinson
Paladin is a PI(4,5)P2 phosphoinositide phosphatase that regulates endosomal signaling and angiogenesis
Anja Nitzsche, Riikka Pietila, Chiara Testini, Takeshi Ninchoji, Ross O Smith, Elisabet Ekvarn, Jimmy Larsson, Francis P Roche, Isabel Egana, Suvi Jauhiainen, Philipp Berger, Lena Claesson-Welsh, Mats Hellstrom
Apical Constriction Reversal upon Mitotic Entry Underlies Different Morphogenetic Outcomes of Cell Division
Clint S. Ko, Prateek Kalakuntla, Adam C. Martin
The Pebble/Rho1/Anillin pathway controls polyploidization and axonal wrapping activity in the glial cells of the Drosophila eye
Lígia Tavares, Patrícia Gracio, Raquel Ramos, Rui Traquete, João B Relvas, Paulo S Pereira
Systematic functional analysis of Rab GTPases reveals limits of neuronal robustness in Drosophila
Friederike E. Kohrs, Ilsa-Maria Daumann, Bojana Pavlović, Eugene Jennifer Jin, Shih-Ching Lin, Fillip Port, F. Ridvan Kiral, Heike Wolfenberg, Thomas F. Mathejczyk, Chih-Chiang Chan, Michael Boutros, P. Robin Hiesinger
Cytoplasmic sharing through apical membrane remodeling
Nora G. Peterson, Benjamin M. Stormo, Kevin P. Schoenfelder, Juliet S. King, Rayson R. S. Lee, Donald T. Fox
The localization of chitin synthase mediates the patterned deposition of chitin in developing Drosophila bristles
Paul N. Adler
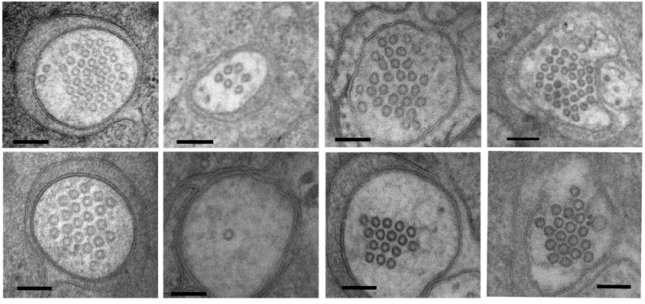
F-box protein MEC-15 promotes microtubule stability and neurite growth by antagonizing the HSP90 chaperone network in Caenorhabditis elegans
Chaogu Zheng, Emily Atlas, Ho Ming Terence Lee, Susan Laura Javier Jao, Ken C. Q. Nguyen, David H. Hall, Martin Chalfie
| Genes & genomes
Jeremy Lotto, Sibyl Drissler, Rebecca Cullum, Wei Wei, Manu Setty, Erin M. Bell, Stéphane C. Boutet, Sonja Nowotschin, Ying-Yi Kuo, Vidur Garg, Dana Pe’er, Deanna M. Church, Anna-Katerina Hadjantonakis, Pamela A. Hoodless
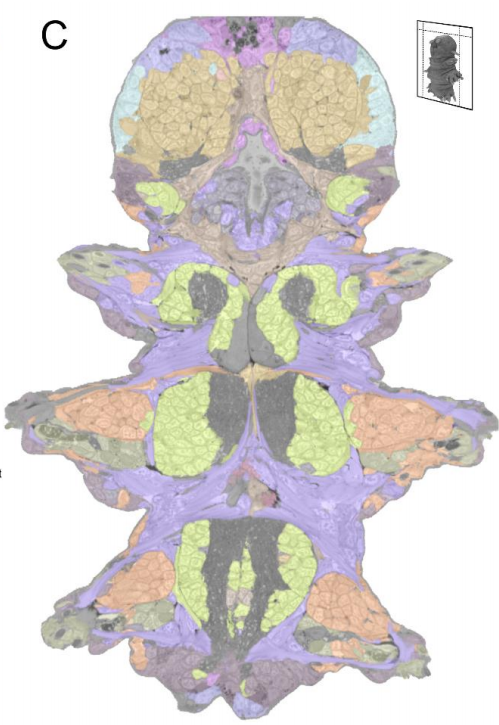
Platynereis gene expression/EM overlay from Vergara et al.
Whole-body integration of gene expression and single-cell morphology
Hernando M. Vergara, Constantin Pape, Kimberly I. Meechan, Valentyna Zinchenko, Christel Genoud, Adrian A. Wanner, Benjamin Titze, Rachel M. Templin, Paola Y. Bertucci, Oleg Simakov, Pedro Machado, Emily L. Savage, Yannick Schwab, Rainer W. Friedrich, Anna Kreshuk, Christian Tischer, Detlev Arendt
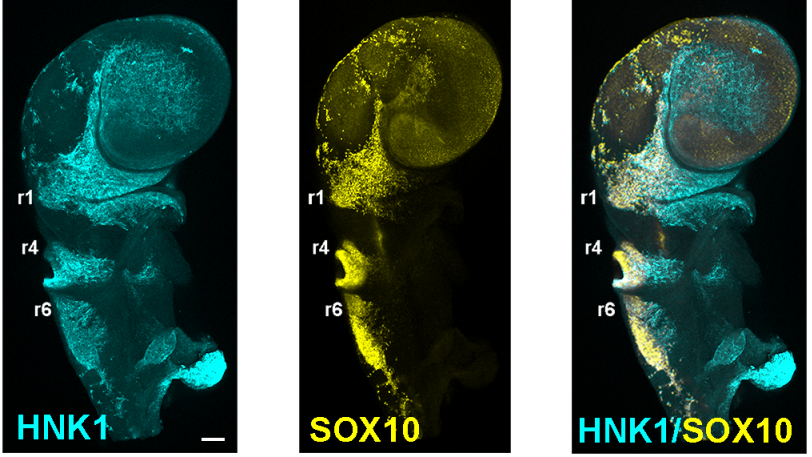
Transcriptome profiling of the branchial arches reveals cell type composition and a conserved signature of neural crest cell invasion
Jason A Morrison, Rebecca McLennan, Jessica M Teddy, Allison R Scott, Jennifer C Kasemeier-Kulesa, Madelaine M Gogol, Paul M Kulesa
Combinatorial action of transcription factors in open chromatin contributes to early cellular heterogeneity and organizer mesendoderm specification
Ann Rose Bright, Siebe van Genesen, Qingqing Li, Simon J. van Heeringen, Alexia Grasso, Gert Jan C. Veenstra
New observations on non-coding RNAs involved in the dual translation system in zebrafish development
Timo M. Breit, Johanna F. B. Pagano, Pjotr L. van der Jagt, Ellis Mittring, Wim A. Ensink, Marina van Olst, Selina van Leeuwen, Wim de Leeuw, Ulrike Nehrdich, Herman P. Spaink, Han Rauwerda, Rob J. Dekker
Maternal- and somatic-type snoRNA expression and processing in zebrafish development
Johanna F.B. Pagano, Mauro D. Locati, Wim A Ensink, Marina van Olst, Selina van Leeuwen, Wim C. De Leeuw, Ulrike Nehrdich, Herman P Spaink, Han Rauwerda, Martijs J. Jonker, Rob J. Dekker, Timo M Breit
Pou5f3 and Sox19b select gene expression repertoire at Zygotic Genome Activation
Meijiang Gao, Marina Veil, Marcus Rosenblatt, Anna Gebhard, Helge Hass, Lenka Buryanova, Lev Y. Yampolsky, Björn Grüning, Jens Timmer, Daria Onichtchouk
Evolutionarily conserved regulation of embryonic fast-twitch skeletal muscle differentiation by Pbx factors
Gist H. Farr III, Bingsi Li, Maurizio Risolino, Nathan M. Johnson, Zizhen Yao, Robert M. Kao, Mark W. Majesky, Stephen J. Tapscott, Licia Selleri, Lisa Maves
Expression of a Barhl1a reporter in subsets of retinal ganglion cells and commissural neurons of the developing zebrafish brain
Shahad Albadri, Olivier Armant, Tairi Aljand-Geschwill, Filippo Del Bene, Matthias Carl, Uwe Straehle, Lucia Poggi
The GINS complex is required for the survival of rapidly proliferating retinal and tectal progenitor cells during zebrafish development
Máté Varga, Kitti Csályi, István Bertyák, Dóra K. Menyhárd, Richard J. Poole, Kara L. Cerveny, Dorottya Kövesdi, Balázs Barátki, Hannah Rouse, Zsuzsa Vad, Thomas A. Hawkins, Heather L. Stickney, Florencia Cavodeassi, Quenten Schwarz, Rodrigo M. Young, Stephen W. Wilson
Hnrnpul1 loss of function affects skeletal and limb development
Danielle L Blackwell, Sherri D Fraser, Oana Caluseriu, Claudia Vivori, Paul MK Gordon, Amanda V Tyndall, Ryan E Lamont, Jillian S Parboosingh, A Micheil Innes, François P Bernier, Sarah J Childs
Sox17 and β-catenin co-occupy Wnt-responsive enhancers to govern the endodermal gene regulatory network
Shreyasi Mukherjee, Praneet Chaturvedi, Scott A Rankin, Margaret B Fish, Marcin Wizla, Kitt D. Paraiso, Melissa MacDonald, Xiaoting Chen, Matthew T. Weirauch, Ira L. Blitz, Ken W.Y. Cho, Aaron Zorn
Characterising open chromatin identifies novel cis-regulatory elements important for paraxial mesoderm formation and axis extension
Gi Fay Mok, Leighton Folkes, Shannon Weldon, Eirini Maniou, Victor Martinez-Heredia, Alice Godden, Ruth Williams, Grant N. Wheeler, Simon Moxon, Andrea E. Münsterberg
3D Epigenomic Characterization Reveals Insights Into Gene Regulation and Lineage Specification During Corticogenesis
Michael Song, Mark-Phillip Pebworth, Xiaoyu Yang, Armen Abnousi, Changxu Fan, Jia Wen, Jonathan D. Rosen, Mayank NK Choudhary, Xiekui Cui, Ian R. Jones, Seth Bergenholtz, Ugomma C. Eze, Ivan Juric, Bingkun Li, Lenka Maliskova, Weifang Liu, Alex A. Pollen, Yun Li, Ting Wang, Ming Hu, Arnold R. Kriegstein, Yin Shen
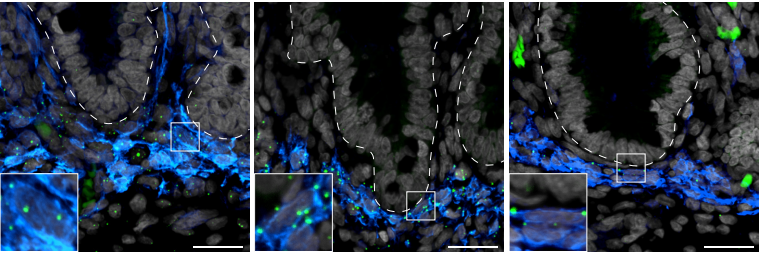
In vitro and in vivo development of the human intestinal niche at single cell resolution
Michael Czerwinski, Emily M. Holloway, Yu-Hwai Tsai, Angeline Wu, Qianhui Yu, Josh Wu, Katherine D. Walton, Caden Sweet, Charlie Childs, Ian Glass, Barbara Treutlein, J. Gray Camp, Jason R. Spence
Single-cell sequencing of developing human gut reveals transcriptional links to childhood Crohn’s disease
Rasa Elmentaite, Alexander Ross, Kylie R. James, Daniel Ortmann, Tomas Gomes, Kenny Roberts, Komal Nayak, Liz Tuck, Omer Ali Bayraktar, Robert Heuschkel, Ludovic Vallier, Sarah A. Teichmann, Matthias Zilbauer
Prediction of meiosis-essential genes based upon the dynamic proteomes responsive to spermatogenesis
Kailun Fang, Qidan Li, Yu Wei, Jiaqi Shen, Wenhui Guo, Changyang Zhou, Ruoxi Wu, Wenqin Ying, Lu Yu, Jin Zi, Yuxing Zhang, Hui Yang, Siqi Liu, Charlie Degui Chen
Cytosine methylation dynamics during post-testicular sperm maturation in mammals
Carolina Galan, Ryan W. Serra, Fengyun Sun, Vera D. Rinaldi, Colin C. Conine, Oliver J. Rando
ATRIP protects progenitor cells against DNA damage in vivo
Gabriel E. Matos-Rodrigues, Paulius Grigaravicius, Bernard S. Lopez, Thomas Hofmann, Pierre-Olivier Frappart, Rodrigo A. P. Martins
A Casz1 – NuRD complex regulates temporal identity transitions in neural progenitors
Pierre Mattar, Christine Jolicoeur, Sujay Shah, Michel Cayouette
CHD4-NURD controls spermatogonia survival and differentiation
Rodrigo O. de Castro, Victor Goitea, Luciana Previato, Agustin Carbajal, Courtney T. Griffin, Roberto J. Pezza
Fine-tuning of the PAX-SIX-EYA-DACH network by multiple microRNAs controls embryo myogenesis
Camille Viaut, Andrea Munsterberg
Sp8 regulatory function in the limb bud ectoderm
Rocío Pérez-Gómez, Marc Fernández-Guerrero, Víctor Campa, Juan F. Lopez-Gimenez, Alvaro Rada-Iglesias, Maria A. Ros
The homeoprotein ENGRAILED-1 promotes motoneuron survival and motor functions
Stephanie E. Vargas Abonce, Mélanie Lebœuf, Kenneth L. Moya, Alain Prochiantz
DUX4 regulates oocyte to embryo transition in human
Sanna Vuoristo, Christel Hydén-Granskog, Masahito Yoshihara, Shruti Bhagat, Lisa Gawriyski, Eeva-Mari Jouhilahti, Anastassius Damdimopoulos, Vipin Ranga, Mahlet Tamirat, Mikko Huhtala, Kosuke Hashimoto, Kaarel Krjutškov, Gaëlle Recher, Sini Ezer, Priit Paluoja, Pauliina Paloviita, Yujiro Takegami, Ai Kanemaru, Karolina Lundin, Tomi Airenne, Timo Otonkoski, Juha S. Tapanainen, Hideya Kawaji, Yasuhiro Murakawa, Thomas R. Bürglin, Markku Varjosalo, Mark S. Johnson, Timo Tuuri, Shintaro Katayama, Juha Kere
Hnf4a-mediated regulation of proximal tubule progenitors in the mouse kidney
Sierra S. Marable, Eunah Chung, Joo-Seop Park
Redundant function of Ets1 and Ets2 in regulating M-phase progression in post-natal angiogenesis
Sankha Ghosh, Catherine B MarElia-Bennett, Blake E Hildreth III, Julia E Lefler, Sudarshana M Sharma, Michael C Ostrowski
Smchd1 is a maternal effect gene required for autosomal imprinting
Iromi Wanigasuriya, Quentin Gouil, Sarah A. Kinkel, Andrés Tapia del Fierro, Tamara Beck, Ellise E.A. Roper, Kelsey Breslin, Jessica Stringer, Karla Hutt, Heather J. Lee, Andrew Keniry, Matthew E. Ritchie, Marnie E. Blewitt
PEA15 loss of function and defective cerebral development in the domestic cat
Emily C. Graff, J. Nicholas Cochran, Christopher B. Kaelin, Kenneth Day, Heather L. Gray-Edwards, Rie Watanabe, Jey W. Koehler, Rebecca A. Falgoust, Jeremy W. Prokop, Richard M. Myers, Nancy R. Cox, Gregory S. Barsh, Douglas R. Martin, 99 Lives Consortium
Hmx3a does not require its homeodomain for its essential functions in spinal cord, ear and lateral line development
Samantha J. England, Gustavo A. Cerda, Angelica Kowalchuk, Taylor Sorice, Ginny Grieb, Katharine E. Lewis
OTX2 non-cell autonomous activity regulates inner retinal function
Raoul Torero-Ibad, Bilal Mahzar, Clémentine Vincent, Clémence Bernard, Julie Dégardin, Manuel Simonutti, Thomas Lamonerie, Ariel Di Nardo, Alain Prochiantz, Kenneth L. Moya
Antisense ncRNAs during early vertebrate development are divided in groups with distinct features
Sanjana Pillay, Hazu
RNA-seq and scRNA-seq reveal trajectory progression of the retinal ganglion cell lineage in wild-type and Atoh7-null retinas
Fuguo Wu, Jonathan E. Bard, Julien Kann, Donald Yergeau, Darshan Sapkota, Yichen Ge, Zihua Hu, Jie Wang, Tao Liu, Xiuqian Mu
Defective heart chamber growth and myofibrillogenesis after knockout of adprhl1 gene function by targeted disruption of the ancestral catalytic active site
Stuart J. Smith, Norma Towers, Kim Demetriou, Timothy J. Mohun
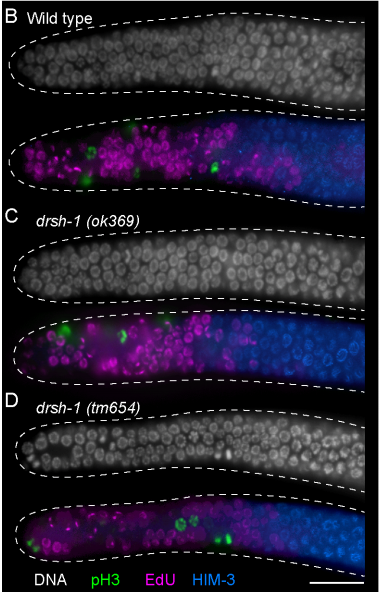
Drosha Regulates Oogenesis and microRNAs Germline Autonomously and Non-autonomously in C. elegans
Amanda L. Minogue, Kenneth A. Trimmer, Jacob H. Seemann, Awdhesh Kalia, Swathi Arur
The predicted RNA-binding protein ETR-1/CELF1 acts in muscles to regulate neuroblast migration in Caenorhabditis elegans
Matthew E. Ochs, Matthew P. Josephson, Erik A. Lundquist
Argonaute catalytic activity is required for maternal mRNA clearance in embryos
Piergiuseppe Quarato, Meetali Singh, Eric Cornes, Blaise Li, Loan Bourdon, Celine Didier, Germano Cecere
C. elegans nuclear RNAi factor SET-32 deposits the transgenerational heritable histone modification, H3K23me3
Lianna Schwartz-Orbach, Chenzhen Zhang, Simone Sidoli, Richa Amin, Diljeet Kaur, Anna Zhebrun, Julie Ni, Sam Guoping Gu
The ancestral C. elegans cuticle suppresses rol-1
Luke M. Noble, Asif Miah, Taniya Kaur, Matthew V. Rockman
The Hox transcription factor Ubx ensures somatic myogenesis by suppressing the mesodermal master regulator Twist
Katrin Domsch, Julia Schröder, Matthias Janeschik, Christoph Schaub, Ingrid Lohmann
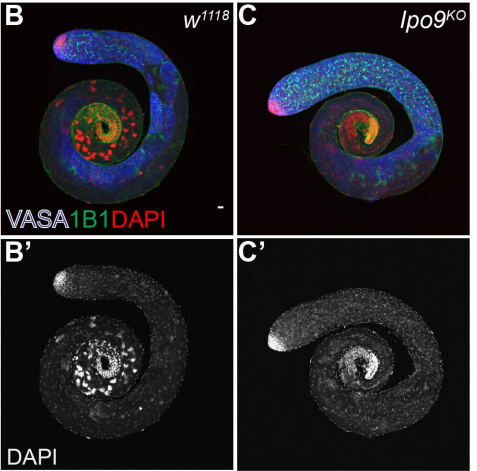
Importin-9 regulates chromosome segregation and packaging in Drosophila germ cells
Victor Palacios, Garrett C Kimble, Tina L. Tootle, Michael Buszczak
Differentiating Drosophila female germ cells initiate Polycomb silencing by altering PRC2 sampling
Steven Z DeLuca, Megha Ghildiyal, Wanbao Niu, Liang-Yu Pang, Allan C. Spradling
The Drosophila HP1 family is associated with active gene expression across chromatin contexts
John M. Schoelz, Justina X. Feng, Nicole C. Riddle
Transcriptome analysis of somatic cell populations in the Drosophila testis links metabolism and stemness
Silvana Hof-Michel, Christian Bökel
Intimate functional interactions between TGS1 and the Smn complex revealed by an analysis of the Drosophila eye development
Paolo Maccallini, Francesca Bavasso, Livia Scatolini, Elisabetta Bucciarelli, Gemma Noviello, Veronica Lisi, Valeria Palumbo, Simone D’Angeli, Stefano Cacchione, Giovanni Cenci, Laura Ciapponi, James G. Wakefield, Maurizio Gatti, Grazia Daniela Raffa
Developmentally-orchestrated mitochondrial processes prime the selection against harmful mtDNA mutations
Zhe Chen, Zong-Heng Wang, Guofeng Zhang, Christopher K. E. Bleck, Dillon J. Chung, Grey Madison, Eric Lindberg, Christian Combs, Robert S. Balaban, Hong Xu
Impact of gestational low-protein intake on embryonic kidney microRNA expression and in the nephron progenitor cells of the male offspring fetus
Letícia de Barros Sene, Íscia Lopes-Cendes, Wellerson Rodrigo Scarano, Adriana Zapparoli, José Antônio Rocha Gontijo, Patrícia Aline Boer
| Stem cells, regeneration & disease modelling
Generation of twenty four induced pluripotent stem cell lines from twenty four members of the Lothian Birth Cohort 1936
Jamie Toombs, Lindsay Panther, Loren Ornelas, Chunyan Liu, Emilda Gomez, Raquel Martín-Ibáñez, Simon R. Cox, Stuart J. Ritchie, Sarah E. Harris, Adele Taylor, Paul Redmond, Tom C. Russ, Lee Murphy, James D. Cooper, Karen Burr, Bhuvaneish T. Selvaraj, Cathy Browne, Clive N. Svendsen, Sally A. Cowley, Ian J. Deary, Siddharthan Chandran, Tara Spires-Jones, Dhruv Sareen
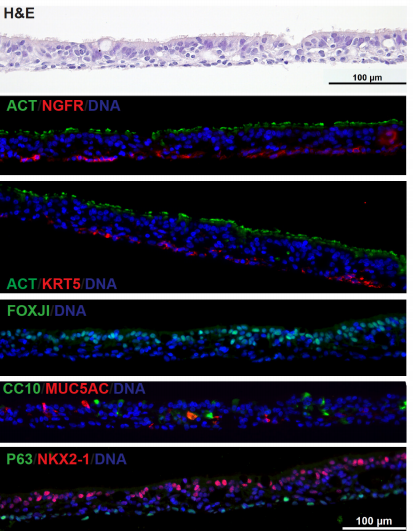
Derivation of Airway Basal Stem Cells from Human Pluripotent Stem Cells
Finn J. Hawkins, Shingo Suzuki, Mary Lou Beermann, Cristina Barillà, Ruobing Wang, Carlos Villacorta-Martin, Andrew Berical, J.C. Jean, Jake Le Suer, Chantelle Simone-Roach, Yang Tang, Thorsten M. Schlaeger, Ana M. Crane, Sarah X. L. Huang, Scott H. Randell, Andras Rab, Eric J. Sorscher, Amjad Horani, Steven L. Brody, Brian R. Davis, Darrell N. Kotton
Profiling of the reprogramome and quantification of fibroblast reprogramming to pluripotency
Kejin Hu, Lara Ianov, David Crossman
Cell-cell communication through FGF4 generates and maintains robust proportions of differentiated cell fates in embryonic stem cells
Dhruv Raina, Angel Stanoev, Azra Bahadori, Michelle Protzek, Aneta Koseska, Christian Schröter
Quantitative and molecular differences distinguish adult human medullary and extramedullary haematopoietic stem and progenitor cell landscapes
Nicole Mende, Hugo Bastos, Antonella Santoro, Kendig Sham, Krishnaa T Mahbubani, Abbie Curd, Hitoshi Takizawa, Nicola K Wilson, Bertie Gottgens, Kourosh Saeb-Parsy, Elisa Laurenti
Variation of human neural stem cells generating organizer states in vitro before committing to cortical excitatory or inhibitory neuronal fates
Nicola Micali, Suel-Kee Kim, Marcelo Diaz-Bustamante, Genevieve Stein-O’Brien, Seungmae Seo, Joo-Heon Shin, Brian G. Rash, Shaojie Ma, Yanhong Wang, Nicolas A. Olivares, Jon Arellano, Kristen R. Maynard, Elana J. Fertig, Alan J. Cross, Roland Burli, Nicholas J. Brandon, Daniel R. Weinberger, Joshua G. Chenoweth, Daniel J. Hoeppner, Nenad Sestan, Pasko Rakic, Carlo Colantuoni, Ronald D. McKay
Differential regulation of lineage commitment in human and mouse primed pluripotent stem cells by NuRD
Ramy Ragheb, Sarah Gharbi, Julie Cramard, Oluwaseun Ogundele, Susan Kloet, Thomas Burgold, Michiel Vermeulen, Nicola Reynolds, Brian Hendrich
Reprogramming of Human Cells to Pluripotency Induces CENP-A Chromatin Depletion
Inês Milagre, Carolina Pereira, Raquel Oliveira, Lars E.T. Jansen
Sequential enrichment at the nuclear periphery of H2A.Zac and H3K9me2 accompanies pluripotency loss in human embryonic stem cells
Georgia Rose Kafer, Regina Rillo-Bohn, Peter M. Carlton
Neural G0: a quiescent-like state found in neuroepithelial-derived cells and glioma
Heather M. Feldman, Chad M. Toledo, Sonali Arora, Pia Hoellerbauer, Philip Corrin, Lucas Carter, Megan Kufeld, Hamid Bolouri, Ryan Basom, Jeffrey Delrow, José L. McFaline-Figueroa, Cole Trapnell, Steven M. Pollard, Anoop Patel, Christopher L. Plaisier, Patrick J. Paddison
TGF-β inhibitor accelerates BMP4-induced cochlear gap junction formation during in vitro differentiation of embryonic stem cells
Ichiro Fukunaga, Cheng Chen, Yoko Oe, Keiko Danzaki, Sayaka Ohta, Akito Koike, Ayumi Fujimoto, Katsuhisa Ikeda, Kazusaku Kamiya
DOT1L Methyltransferase Activity Preserves SOX2-Enhancer Accessibility And Prevents Activation of Repressed Genes In Murine Stem Cells
F. Ferrari, L. Arrigoni, H. Franz, L. Butenko, E. Trompouki, T. Vogel, T. Manke
Trophectoderm Potency is Retained Exclusively in Human Naïve Cells
Ge Guo, Giuliano Giuseppe Stirparo, Stanley Strawbridge, Jian Yang, James Clarke, Meng Amy Li, Sam Myers, Buse Nurten Özel, Jennifer Nichols, Austin Smith
Continual inactivation of genes involved in stem cell functional identity stabilizes progenitor commitment
Noemi Quinto-Rives, Hideyuki Komori, Derek H. Janssens, Shu Kondo, Qi Dai, Adrian W. Moore, Cheng-Yu Lee
MicroRNA-dependent inhibition of PFN2 orchestrates ERK activation and pluripotent state transitions by regulating endocytosis
Carolyn Sangokoya, Robert Blelloch
iPSC-Derived Ovarian Tissue Restores Ovarian Function in Subfertile Mice and After Gonadotoxic Chemotherapy
K.M. Elias, N.W. Ng, K.U. Dam, A. Milne, E.R. Disler, A. Gockley, N. Holub, G.M. Church, E.S. Ginsburg, R.M. Anchan
Cryopreservation of midbrain dopaminergic neural cells differentiated from human embryonic stem cells
Nicola J Drummond, Karamjit Singh Dolt, Maurice A Canham, Peter Kilbride, George John Morris, Tilo Kunath
PDH Mediated Mitochondrial Respiration Controls the Speed of Muscle Stem Cell Activation in Muscle Repair and Aging
Manmeet H. Raval, Pin-Chung Cheng, Nicholas Guardino, Sanjana Ahsan, Hao Zhou, Rajiv Lochan Tiwari, Lu Wang, Andrew Chareunsouk, Maxwell Ederer, Ara B. Hwang, Matt Ellenberger, Rob Pepin, Daniel Raftery, Daniel Promislow, Keyue Shen, Andrew S. Brack, Joseph T. Rodgers
Accelerated cell cycles enable organ regeneration under developmental time constraints in the Drosophila hindgut
Erez Cohen, Donald T. Fox
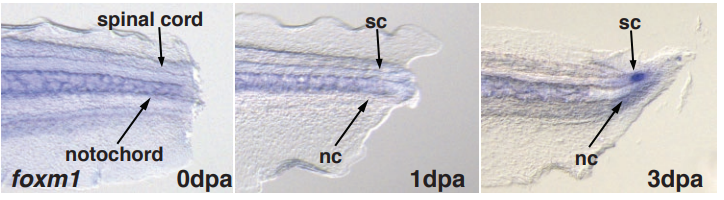
Foxm1 regulates neuronal progenitor fate during spinal cord regeneration
Diane Pelzer, Lauren S. Phipps, Raphael Thuret, Syed Murtuza Baker, Karel Dorey

Suppression of Inflammation Delays Hair Cell Regeneration and Functional Recovery Following Lateral Line Damage in Zebrafish Larvae
Ru Zhang, Xiao-Peng Liu, Ya-Juan Li, Ming Wang, Lin Chen, Bing Hu
Localized activation of ependymal progenitors induces EMT-mediated glial bridging after spinal cord injury
Lili Zhou, Brooke Burris, Ryan Mcadow, Mayssa H Mokalled
Epigenetic immune-modulation by Histone Deacetylase Activity (HDAC) of tissue and organ regeneration in Xenopsu laevis
Nathalia Pentagna, Felipe Soares dos Santos, Fernanda Martins de Almeida, José Garcia Abreu, Michael Levin, Katia Carneiro
Ultrafiltration Segregates Tissue Regenerative Stimuli Harboured Within and Independent of Extracellular Vesicles
TT Cooper, SE Sherman, T Dayarathna, GI Bell, Jun Ma, DM McRae, F Lagugné-Labarthet, SH Pasternak, GA Lajoie, DA Hess
Expression of dlx genes in the normal and regenerating brain of adult zebrafish.
Hellen Weinschutz Mendes, Mariam TAKTEK, Thomas DURET, Marc Ekker
Arid1a loss potentiates pancreatic β-cell regeneration through activation of EGF signaling
Cemre Celen, Jen-Chieh Chuang, Shunli Shen, Jordan E. Otto, Clayton K. Collings, Xin Luo, Lin Li, Yunguan Wang, Zixi Wang, Yuemeng Jia, Xuxu Sun, Ibrahim Nassour, Jiyoung Park, Alexandra Ghaben, Tao Wang, Sam C. Wang, Philipp E. Scherer, Cigall Kadoch, Hao Zhu
ARHGEF3 regulates skeletal muscle regeneration and strength through autophagy
Jae-Sung You, Nilmani Singh, Adriana Reyes-Ordonez, Nidhi Khanna, Zehua Bao, Huimin Zhao, Jie Chen
Cerebral organoid model reveals excessive proliferation of human caudal late interneuron progenitors in Tuberous Sclerosis Complex
Oliver L. Eichmüller, Nina S. Corsini, Ábel Vértesy, Theresa Scholl, Victoria-Elisabeth Gruber, Angela M. Peer, Julia Chu, Maria Novatchkova, Mercedes F. Paredes, Martha Feucht, Jürgen A. Knoblich
Altered patterning of interneuron progenitors in Down syndrome
Yathinder Giffin-Rao, Bennett Strand, Margaret Medo, Aratrika Keshan, Roger A. Daley Jr., Sruti Mohan, Samuel Dantienne, Bradley Levesque, Lindsey Amundson, Leslie Huang, Rebecca Reese, Daifeng Wang, Su-Chun Zhang, Anita Bhattacharyya
Lack of dcf1 leads to neuronal migration delay, axonal swollen and autism-related deficits
Ruili Feng, Yanlu Chen, Yangyang Sun, Guanghong Luo, Jianjian Guo, Qiang Liu, Jie Wu, Xiangchun Ju, Tieqiao Wen
Proteomic and phosphoproteomic analysis identifies novel liver-related signaling in retinal pigment epithelial cells during epithelial-mesenchymal transition
Joseph L. Mertz, Srinivas Sripathi, X
Direct reprogramming of astrocytes to neurons leads to functional recovery after stroke
Jessica Livingston, Tina Lee, Emerson Daniele, Clara Phillips, Alexandra Krassikova, Tom Enbar, Ines Kortebi, K.W. Annie Bang, Brennan Donville, Omadyor Ibragimov, Nadia Sachewsky, Cindi M Morshead, Maryam Faiz
Alzheimer’s disease-relevant tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model
Sanjib Guha, Sarah Fischer, Gail VW Johnson, Keith Nehrke
Single Cell Transcriptomics Reveals Dysregulated Cellular and Molecular Networks in a Fragile X Syndrome model
Elisa Donnard, Huan Shu, Manuel Garber
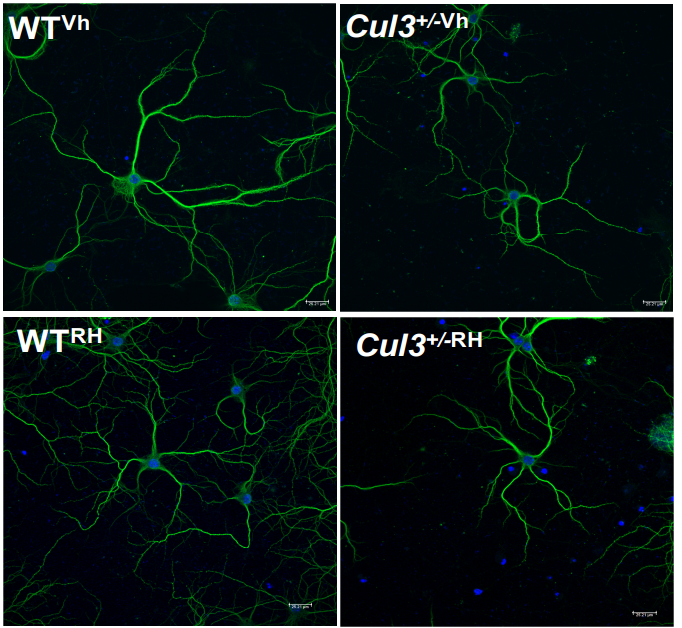
Autism-linked Cullin3 germline haploinsufficiency impacts cytoskeletal dynamics and cortical neurogenesis through RhoA signaling
Megha Amar, Akula Bala Pramod, Victor Munive Herrera, Nam-Kyung Yu, Lily R Qiu, Pan Zhang, Patricia Moran-Losada, Cleber A Trujillo, Jacob Ellegood, Jorge Urresti, Kevin Chau, Jolene Diedrich, Jiaye Chen, Jessica Gutierrez, Jonathan Sebat, Dhakshin Ramanathan, Jason P Lerch, John R Yates III, Alysson R Muotri, Lilia M Iakoucheva
Loss of epithelial polarity redirects Notch signaling and triggers a Xrp1 response during neoplastic growth in Drosophila
Rémi Logeay, Charles Géminard, Patrice Lassus, Diala Kantar, Lisa Héron-Milhavet, Bettina Fischer, Sarah J. Bray, Jacques Colinge, Alexandre Djiane
Genome-wide molecular effects of the neuropsychiatric 16p11 CNVs in an iPSC-to-iN neuronal model
Thomas R. Ward, Xianglong Zhang, Louis C. Leung, Bo Zhou, Kristin Muench, Julien G. Roth, Arineh Khechaduri, Melanie J. Plastini, Carol Charlton, Reenal Pattni, Steve Ho, Marcus Ho, Yiling Huang, Joachim F. Hallmayer, Phillippe Mourrain, Theo D. Palmer, Alexander E. Urban
ASH1L REGULATES THE STRUCTURAL DEVELOPMENT OF NEURONAL CIRCUITRY BY MODULATING BDNF/TrkB SIGNALING IN HUMAN NEURONS
Seon H Cheon, Allison M Culver, Anna M Bagnell, Foster D Ritchie, Janay M Clytus, Mykayla McCord, Carin M Pappendorp, Evelyn Chukwurah, Austin J Smith, Mara H Cowen, Pankaj S Ghate, Shannon W Davis, Judy S Liu, Sofia Lizarraga
Successful Correction of ALD Patient-derived iPSCs Using CRISPR/Cas9
Eul Sik Jung, Zhejiu Quan, Mi-Yoon Chang, Wonjun Hong, Ji Hun Kim, Seung Hyun Kim, Seungkwon You, Dae-Sung Kim, Jiho Jang, Sang-Hun Lee, Hyongbum (Henry) Kim, Hoon Chul Kang
| Plant development
Hook shape of growing leaves results from an active regulation
Mathieu Rivière, Yoann Corre, Alexis Peaucelle, Julien Derr, Stéphane Douady
Co-expression clustering across flower development identifies modules for diverse floral forms in Achimenes (Gesneriaceae)
Wade R Roberts, Eric H Roalson
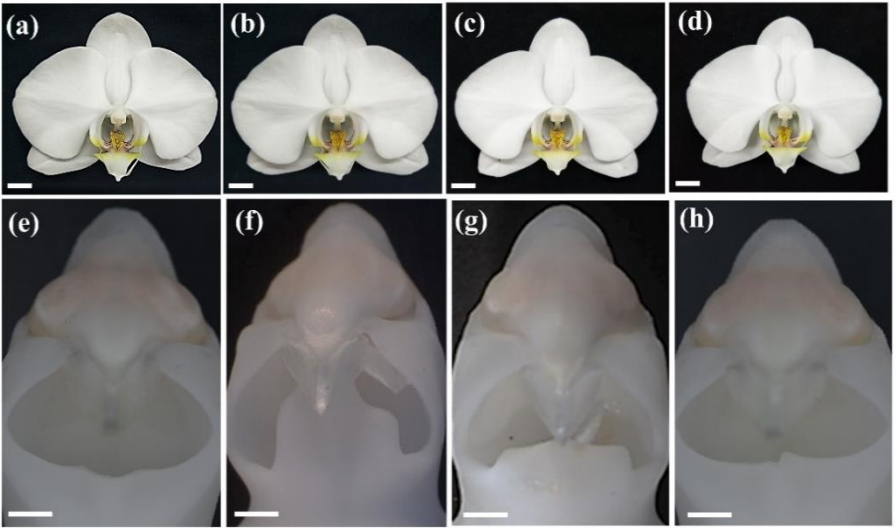
Ancestral duplicated DL/CRC orthologs display function on orchid reproductive organ innovation
You-Yi Chen, Yu-Yun Hsiao, Chung-I Li, Chuan-Ming Yeh, Nobutaka Mitsuda, Hong-xing Yang, Chi-Chou Chiu, Song-Bin Chang, Zhong-Jian Liu, Wen-Chieh Tsai
Protein complex stoichiometry and expression dynamics of transcription factors modulate stem cell division
Natalie M. Clark, Adam P. Fisher, Barbara Berckmans, Lisa Van den Broeck, Emily C. Nelson, Thomas T. Nguyen, Estefano Bustillo-Avendaño, Sophia G. Zebell, Miguel Moreno-Risueno, Rüdiger Simon, Kimberly L. Gallagher, Rosangela Sozzani
ERECTA family signaling constrains CLAVATA3 and WUSCHEL to the center of the shoot apical meristem
Liang Zhang, Daniel DeGennaro, Guangzhong Lin, Jijie Chai, Elena D. Shpak
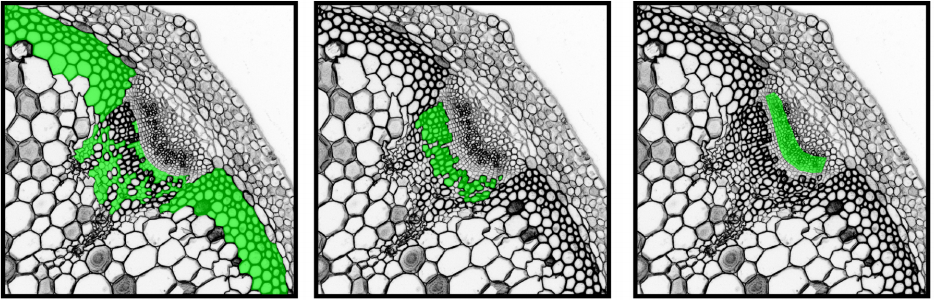
Tissue-specific transcriptome profiling of the Arabidopsis thaliana inflorescence stem reveals local cellular signatures.
Dongbo Shi, Virginie Jouannet, Javier Agusti, Verena Kaul, Victor Levitsky, Pablo Sanchez, Victoria V Mironova, Thomas Greb
A non-canonical histone acetyltransferase targets intragenic enhancers and regulates plant architecture
Xueyong Yang, Jianbin Yan, Zhen Zhang, Tao Lin, Tongxu Xin, Bowen Wang, Shenhao Wang, Jicheng Zhao, Zhonghua Zhang, William J. Lucas, Guohong Li, Sanwen Huang
Global Dynamic Molecular Profiles of Stomatal Lineage Cell Development by Single-Cell RNA Sequencing
Zhixin Liu, Yaping Zhou, Jinggong Guo, Jiaoai Li, Zixia Tian, Zhinan Zhu, Jiajing Wang, Rui Wu, Bo Zhang, Yongjian Hu, Yijing Sun, Yan Shangguan, Weiqiang Li, Tao Li, Yunhe Hu, Chenxi Guo, Jean-David Rochaix, Yuchen Miao, Xuwu Sun
DRT111/SFPS splicing factor controls ABA sensitivity in Arabidopsis seed development and germination
Paola Punzo, Alessandra Ruggiero, Marco Possenti, Giorgio Perrella, Roberta Nurcato, Antonello Costa, Giorgio Morelli, Stefania Grillo, Giorgia Batelli
GSK3 activity is a cell fate switch that balances the ratio of vascular cell type
Takayuki Tamaki, Satoyo Oya, Makiko Naito, Yasuko Ozawa, Tomoyuki Furuya, Masato Saito, Mayuko Sato, Mayumi Wakazaki, Kiminori Toyooka, Hiroo Fukuda, Ykä Helariutta, Yuki Kondo
Vegetative nuclear positioning is required for calcium and ROS signaling in Arabidopsis pollen tubes
Morgan Moser, Andrew Kirkpatrick, Norman Reid Groves, Iris Meier
Perturbation of GABA Biosynthesis Links Cell Cycle to Control Arabidopsis thaliana Leaf Development
Yaxin Gong, Han Yue, Yu Xiang, Guanghui Yu
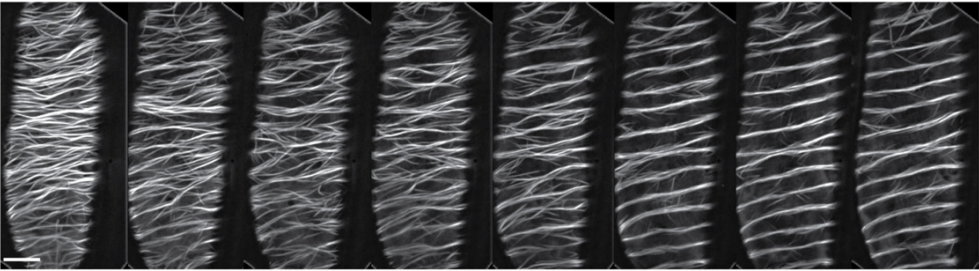
Long-term single-cell imaging and simulations of microtubules reveal driving forces for wall pattering during proto-xylem development
René Schneider, Kris van ’t Klooster, Kelsey Picard, Jasper van der Gucht, Taku Demura, Marcel Janson, Arun Sampathkumar, Eva E. Deinum, Tijs Ketelaar, Staffan Persson
The Arabidopsis gene RGO mediates cytokinin responses and increases seed yield
Jhadeswar Murmu, Ghislaine Allard, Denise Chabot, Eiji Nambara, Raju Datla, Shelley Hepworth, Rajagopal Subramaniam, Jas Singh
SPAs promote thermomorphogenesis via regulating the phyB-PIF4 module in Arabidopsis
Sanghwa Lee, Inyup Paik, Enamul Huq
Genomic evidence reveals SPA-regulated developmental and metabolic pathways in dark-grown Arabidopsis seedlings
Vinh Ngoc Pham, Inyup Paik, Ute Hoecker, Enamul Huq
Class III peroxidases PRX01, PRX44, and PRX73 potentially target extensins during root hair growth in Arabidopsis thaliana
Eliana Marzol, Cecilia Borassi, Philippe Ranocha, Ariel. A. Aptekman, Mauro Bringas, Janice Pennington, Julio Paez-Valencia, Javier Martínez Pacheco, Diana Rosa Rodríguez Garcia, Yossmayer del Carmen Rondón Guerrero, Mariana Carignani, Silvina Mangano, Margaret Fleming, John W. Mishler-Elmore, Francisca Blanco-Herrera, Patricia Bedinger, Christophe Dunand, Luciana Capece, Alejandro D. Nadra, Michael Held, Marisa Otegui, José M. Estevez
Golgi-localized exo-β1,3-galactosidases involved in AGP modification and root cell expansion in Arabidopsis
Pieter Nibbering, Bent L. Petersen, Mohammed Saddik Motawia, Bodil Jørgensen, Peter Ulvskov, Totte Niittylä
Arabidopsis AZG2, an auxin induced putative cytokinin transporter, regulates lateral root emergence
Tomás M. Tessi, Sabine Brumm, Eva Winklbauer, Benjamin Schumacher, Carlos I. Lescano, Claudio A. González, Dierk Wanke, Verónica G. Maurino, Klaus Harter, Marcelo Desimone
Plastid EF-Tu Regulates Root Development through Both the ATM Pathway and GUN1
Pengcheng Li, Junjie Ma, Xueping Sun, Chuanzhi Zhao, Changle Ma, Xingjun Wang
Retrograde induction of phyB orchestrates ethylene-auxin hierarchy to regulate growth
Jishan Jiang, Yanmei Xiao, Wei Hu, Hao Chen, Liping Zeng, Haiyan Ke, Franck A. Ditengou, Upendra Devisetty, Klaus Palme, Julin Maloof, Katayoon Dehesh
KIX8 and KIX9 are conserved repressors of organ size in the asterid species tomato
Gwen Swinnen, Alexandra Baekelandt, Rebecca De Clercq, Jan Van Doorsselaere, Nathalie Gonzalez, Dirk Inzé, Alain Goossens, Laurens Pauwels
The FUSED LEAVES1/ADHERENT1 Regulatory Module Is Required For Maize Cuticle Development And Organ Separation
Xue Liu, Richard Bourgault, Josh Strable, Mary Galli, Zongliang Chen, Jiaqiang Dong, Isabel Molina, Andrea Gallavotti
Evo-devo & evo
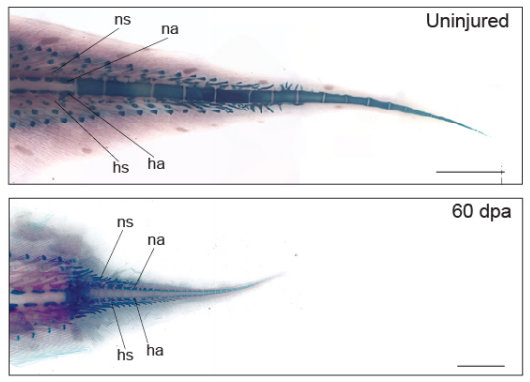
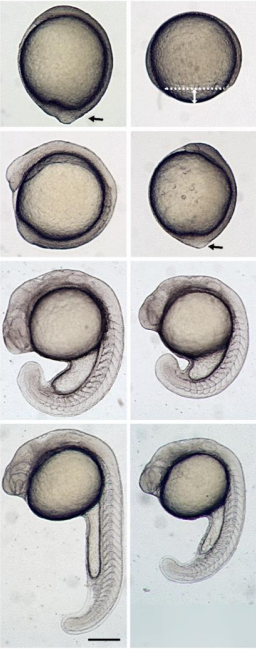
Lungfish tails from Verissimo, et al.
The West African lungfish provides insights into the evolution of tetrapod tail regeneration
Kellen Matos Verissimo, Louise Neiva Perez, Aline Cutrim Dragalzew, Gayani Senevirathne, Sylvain Darnet, Wainna Renata Barroso Mendes, Ciro Ariel dos Santos Neves, Erika Monteiro dos Santos, Cassia Nazare de Sousa Moraes, Neil Shubin, Nadia Belinda Frobisch, Josane de Freitas Sousa, Igor Schneider
Multi-organ transcriptomic landscape of Ambystoma velasci metamorphosis
Palacios-Martínez Janet, Caballero-Pérez Juan, Espinal-Centeno Annie, Marquez-Chavoya Gilberto, Lomelí Hilda, Salas-Vidal Enrique, Schnabel Denhi, Chimal-Monroy Jesus, Cruz-Ramírez Alfredo
Bicaudal C is required for the function of the follicular epithelium during oogenesis in Rhodnius prolixus
Agustina Pascual, Emiliano S. Vilardo, Catalina Taibo, Julia Sabio y García, Rolando Rivera Pomar
Unravelling the developmental and functional significance of an ancient Argonaute duplication
Arie Fridrich, Vengamanaidu Modepalli, Magda Lewandowska, Reuven Aharoni, Yehu Moran
Comparative transcriptomics across nematode life cycles reveal gene expression conservation and correlated evolution in adjacent developmental stages
Min R. Lu, Cheng-Kuo Lai, Ben-Yang Liao, Isheng Jason Tsai
MicroRNA clusters integrate evolutionary constraints on expression and target affinities: the miR-6/5/4/286/3/309 cluster in Drosophila leg development
Zhe Qu, Wing Chung Yiu, Ho Yin Yip, Wenyan Nong, Clare W.C. Yu, Ivy H.T. Lee, Annette Y.P. Wong, Nicola W.Y. Wong, Fiona K.M. Cheung, Ting Fung Chan, Kwok Fai Lau, Silin Zhong, Ka Hou Chu, Stephen S. Tobe, David E.K. Ferrier, William G. Bendena, Jerome H.L. Hui
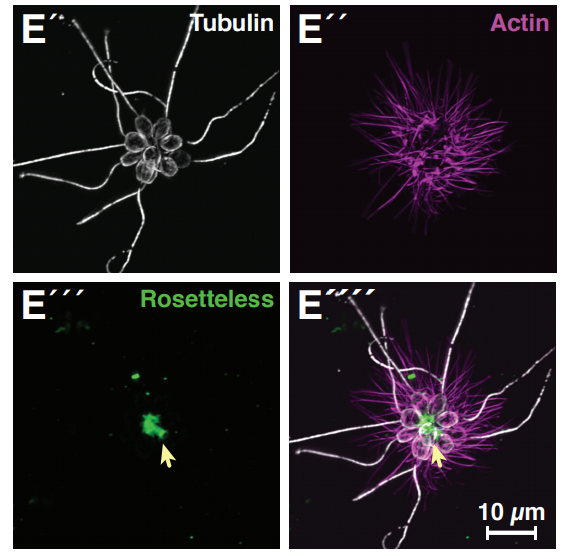
Genome editing enables reverse genetics of multicellular development in the choanoflagellate Salpingoeca rosetta
David S Booth, Nicole King
Integrin-mediated adhesion in the unicellular holozoan Capsaspora owczarzaki
Helena Parra-Acero, Matija Harcet, Núria Sánchez-Pons, Elena Casacuberta, Nicholas H. Brown, Omaya Dudin, Iñaki Ruiz-Trillo
Meta-population structure and the evolutionary transition to multicellularity
Caroline J Rose, Katrin Hammerschmidt, Yuriy Pichugin, Paul B Rainey
The genomic basis of animal origins: a chromosomal perspective from the sponge Ephydatia muelleri
Nathan James Kenny, Warren R Francis, Ramón E Rivera-Vicéns, Ksenia Juravel, Alex de Mendoza, Cristina Díez-Vives, Ryan Lister, Luis Bezares-Calderon, Lauren Grombacher, Maša Roller, Lael D Barlow, Sara Camilli, Joseph F Ryan, Gert Wörheide, April L Hill, Ana Riesgo, Sally Leys
Tracing the origin of a new organ by inferring the genetic basis of rumen evolution
Xiangyu Pan, Yu Wang, Zongjun Li, Xianqing Chen, Rasmus Heller, Nini Wang, Chen Zhao, Yudong Cai, Han Xu, Songhai Li, Ming Li, Cunyuan Li, Shengwei Hu, Hui Li, Kun Wang, Lei Chen, Bin Wei, Zhuqing Zheng, Weiwei Fu, Yue Yang, Tingting Zhang, Zhuoting Hou, Yueyang Yan, Xiaoyang Lv, Wei Sun, Xinyu Li, Shisheng Huang, Lixiang Liu, Shengyong Mao, Wenqing Liu, Jinlian Hua, Zhipeng Li, Guojie Zhang, Yulin Chen, Xihong Wang, Qiang Qiu, Brian P Dalrymple, Wen Wang, Yu Jiang
Cooption of polyalanine tract into a repressor domain in the mammalian transcription factor HoxA11
Vincent J Lynch, Gunter Wagner
Long-term experimental evolution reveals purifying selection on piRNA-mediated control of transposable element expression
Ulfar Bergthorsson, Caroline J. Sheeba, Anke Konrad, Tony Belicard, Toni Beltran, Vaishali Katju, Peter Sarkies
Fly wing evolution explained by a neutral model with mutational pleiotropy
Daohan Jiang, Jianzhi George Zhang
Parent of origin gene expression in the bumblebee, Bombus terrestris, supports Haig’s kinship theory for the evolution of genomic imprinting.
Hollie Marshall, Jelle S van Zweden, Anneleen Van Geystelen, Kristof Benaets, Felix Wäckers, Eamonn B Mallon, Tom Wenseleers
Trans-generational effect of protein restricted diet on adult body and wing size of Drosophila melanogaster
Sudhakar Krittika, Pankaj Yadav
Sex-specific transgenerational plasticity in threespined sticklebacks
Jennifer K Hellmann, Syed Abbas Bukhari, Jack Deno, Alison M Bell
Grandpaternal effects are lineage- and sex-specific in threespined sticklebacks
Jennifer K Hellmann, Erika R Carlson, Alison M Bell
Cell biology
EXOSC10 sculpts the transcriptome during the growth-to-maturation transition in mouse oocytes
Di Wu, Jurrien Dean
A meiosis-specific factor MRM/C19orf57 modulates localization of RAD51 and DMC1 recombinases to DSBs in mouse meiotic recombination
Kazumasa Takemoto, Naoki Tani, Yuki Takada, Sayoko Fujimura, Nobuhiro Tanno, Mariko Yamane, Kaho Okamura, Michihiko Sugimoto, Kimi Araki, Kei-ichiro Ishiguro

The interaction of crossover formation and the dynamic architecture of the synaptonemal complex during meiosis
Simone Köhler, Michal Wojcik, Ke Xu, Abby F. Dernburg
Vps54 regulates Drosophila neuromuscular junction development and controls postsynaptic density composition via a Rab7-dependent mechanism
Prajal H. Patel, Emily C. Wilkinson, Emily L. Starke, Malea R. McGimsey, J. Todd Blankenship, Scott A. Barbee
Transcytosis via the late endocytic pathway as a cell morphogenetic mechanism
Renjith Mathew, Luis Daniel Rios-Barrera, Pedro Machado, Yannick Schwab, Maria Leptin
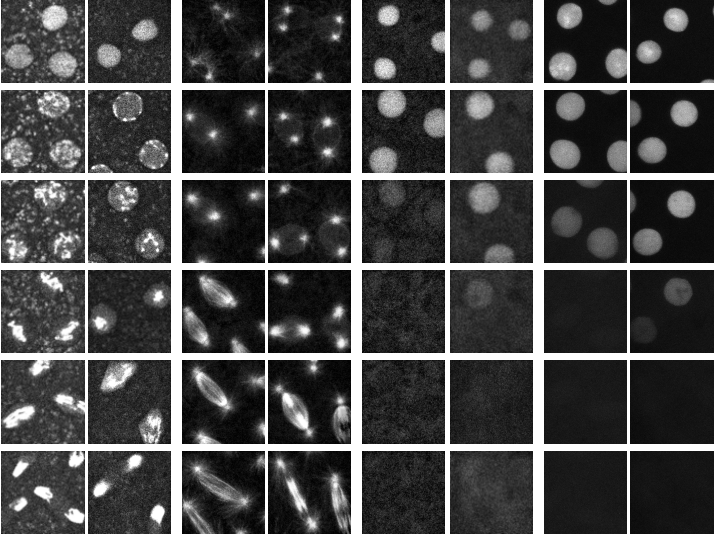
Temperature-Induced Uncoupling of Cell Cycle Regulators
Hanieh Falahati, Woonyung Hur, Stefano Di Talia, Eric F. Wieschaus
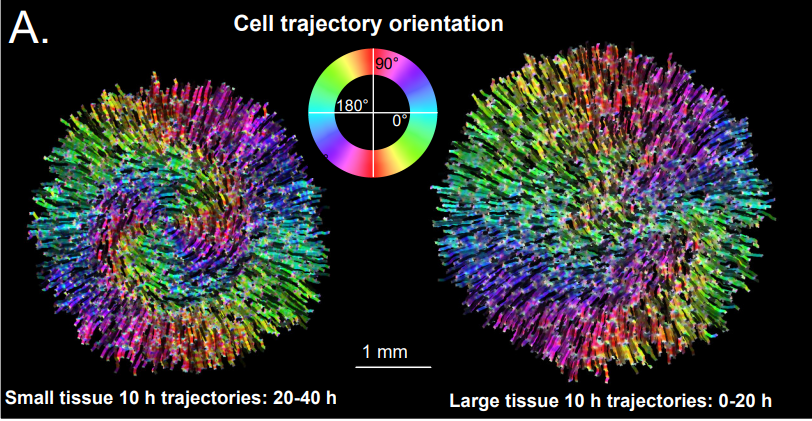
Tissue size controls patterns of cell proliferation and migration in freely-expanding epithelia
Matthew A Heinrich, Julienne LaChance, Tom J. Zajdel, Ricard Alert, Andrej Kosmrlj, Daniel J. Cohen
Vezatin is required for the retrograde axonal transport of endosomes in Drosophila and zebrafish
Michael A. Spinner, Katherine Pinter, Catherine M. Drerup, Tory G. Herman
A paralog-specific role of the COPI pathway in the neuronal differentiation of murine pluripotent cells
Manu Goyal, Xiyan Zhao, Mariya Bozhinova, Karla Lisette Andrade López, Cecilia de Heus, Sandra Schulze-Dramac, Michaela Müller-McNicoll, Judith Klumperman, Julien Béthune
Lamellipodin tunes cell migration by stabilizing protrusions and promoting adhesion formation
Georgi Dimchev, Behnam Amiri, Ashley C. Humphries, Matthias Schaks, Vanessa Dimchev, Theresia E.B. Stradal, Jan Faix, Matthias Krause, Michael Way, Martin Falcke, Klemens Rottner
Microtubule Assembly and Pole Coalescence: Early Steps in C. elegans Oocyte Meiosis I Spindle Assembly
Chien-Hui Chuang, Aleesa J. Schlientz, Jie Yang, Bruce Bowerman
Suppression of canonical TGF-β signaling enables GATA4 to interact with H3K27me3 demethylase JMJD3 to promote cardiomyogenesis
Andrew S Riching, Etienne Danis, Yuanbiao Zhao, Yingqiong Cao, COngwu Chi, Rushita Bagchi, Brianna Klein, Hongyan Xu, TATIANA Kutateladze, Timothy McKinsey, Peter Buttrick, Kunhua Song
Stress fibers are embedded in a contractile cortical network.
Timothee Vignaud, Calina Copos, Christophe Leterrier, Qingzong Tseng, Laurent Blanchoin, Alex Mogilner, Manuel THERY, Laetitia Kurzawa
Integrin Affinity Modulation Critically Regulates Atherogenic Endothelial Activation in vitro and in vivo
Zaki Al-Yafeai, Jonette M. Peretik, Brenna H. Pearson, Umesh Bhattarai, Dongdong Wang, Brian G. Petrich, A. Wayne Orr
Changes in subcellular structures and states of Pumilio1 regulate the translation of target Mad2 and Cyclin B1 mRNAs
Natsumi Takei, Yuki Takada, Shohei Kawamura, Atsushi Saitoh, Jenny Bormann, Wai Shan Yuen, John Carroll, Tomoya Kotani
Leading-edge VASP clusters assemble at sites containing lamellipodin and exhibit size-dependent instability
Karen W. Cheng, R. Dyche Mullins
A non-canonical Hippo pathway regulates spindle disassembly and cytokinesis during meiosis in Saccharomyces cerevisiae
Scott M. Paulissen, Cindy A. Hunt, Christian J. Slubowski, Yao Yu, Dang Truong, Xheni Mucelli, Hung T. Nguyen, Shayla Newman-Toledo, Aaron M. Neiman, Linda S. Huang
Slow Axonal Transport and Presynaptic Targeting of Clathrin Packets
Archan Ganguly, Florian Wernert, Sébastien Phan, Daniela Boassa, Utpal Das, Rohan Sharma, Ghislaine Caillol, Xuemei Han, John R. Yates III, Mark H. Ellisman, Christophe Leterrier, Subhojit Roy
Ctdnep1 and Eps8L2 regulate dorsal actin cables for nuclear positioning during cell migration
Francisco J. Calero-Cuenca, Daniel S. Osorio, Sreerama Chaitanya Sridhara, Yue Jiao, Jheimmy Diaz, Sofia Carvalho-Marques, Bruno Cadot, Edgar R. Gomes
Modelling
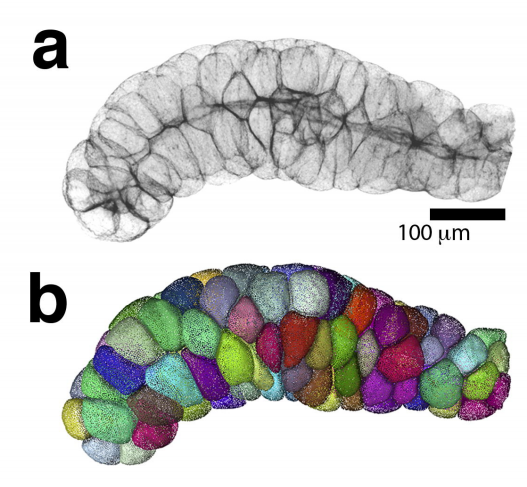
A quantitative principle to understand 3D cellular connectivity in epithelial tubes
Pedro Gomez-Galvez, Pablo Vicente-Munuera, Samira Anbari, Antonio Tagua, Carmen Gordillo, Ana Maria Palacios, Antonio Velasco, Carlos Capitan-Agudo, Clara Grima, Valentina Annese, Rafael Robles, Alberto Marquez, Javier Buceta, Luis M. Escudero
Control of tissue development by cell cycle dependent transcriptional filtering
Maria Abou Chakra, Ruth Isserlin, Thinh Tran, Gary D. Bader
On the preservation of vessel bifurcations during flow-mediated angiogenic remodelling
Lowell T. Edgar, Claudio A. Franco, Holger Gerhardt, Miguel O. Bernabeu
Evolution of multicellularity by collective integration of spatial information
Enrico Sandro Colizzi, Renske M.A. Vroomans, Roeland M.H. Merks
DNA Torsion-based Model of Cell Fate Phase Transitions
Ng Shyh-Chang, Liaofu Luo
Stem cell lineage survival as a noisy competition for niche access
Bernat Corominas-Murtra, Colinda L.G.J. Scheele, Kasumi Kishi, Saskia I.J. Ellenbroek, Benjamin D. Simons, Jacco van Rheenen, Edouard Hannezo
Shape changes and elastic dewetting of adherent epithelia
Benjamin Loewe, Francesco Serafin, Suraj Shankar, Mark J. Bowick, M. Cristina Marchetti
Controlled neighbor exchanges drive glassy behavior, intermittency and cell streaming in epithelial tissues
Amit Das, Srikanth Sastry, Dapeng Bi
Identifying density-dependent interactions in collective cell behaviour
Alexander P Browning, Wang Jin, Michael J Plank, Matthew J Simpson
Tools & resources
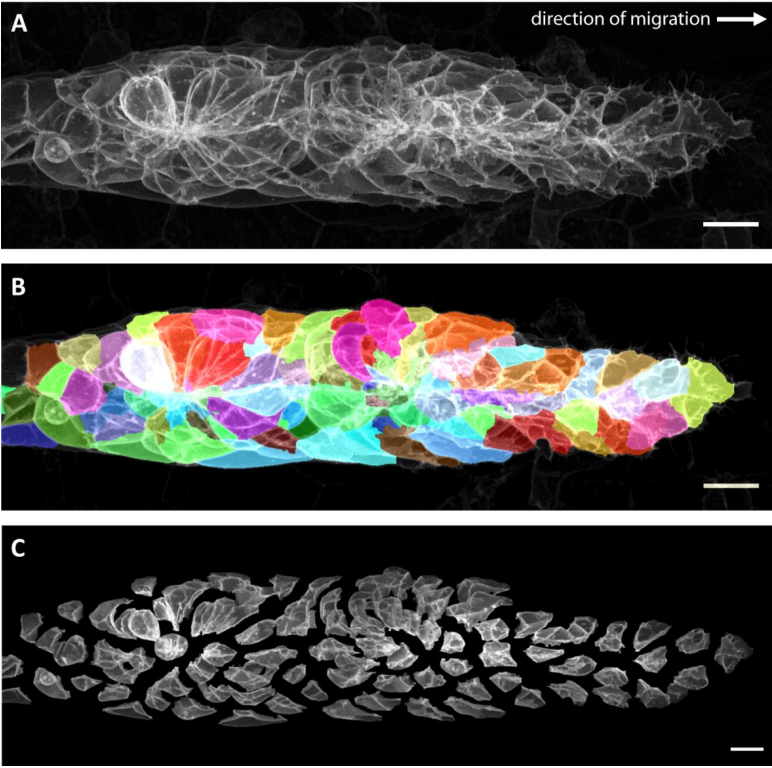
Cell segmentation from Hartmann, et al.
An Image-Based Data-Driven Analysis of Cellular Architecture in a Developing Tissue
Jonas Hartmann, Mie Wong, Elisa Gallo, Darren Gilmour
Capybara: A computational tool to measure cell identity and fate transitions
Wenjun Kong, Yuheng C. Fu, Samantha A. Morris
ZipSeq : Barcoding for Real-time Mapping of Single Cell Transcriptomes
Kenneth H. Hu, John P. Eichorst, Chris S. McGinnis, David M. Patterson, Eric D. Chow, Kelly Kersten, Stephen C. Jameson, Zev J. Gartner, Arjun A. Rao, Matthew F. Krummel
Simultaneous Profiling of DNA Copy Number Variations and Transcriptional Programs in Single Cells using RNA-seq
Ali Madipour-Shirayeh, Natalie Erdmann, Chungyee Leung-Hagesteijn, Paola Neri, Ines Tagoug, Rodger E. Tiedemann
Next-generation cytosine base editors with minimized unguided DNA and RNA off-target events and high on-target activity
Yi Yu, Thomas Leete, David A Born, Lauren Young, Luis A Barrera, Seung-Joo Lee, Holly A Rees, Giuseppe Ciaramella, Nicole M Gaudelli
Multiplexed conditional genome editing with Cas12a in Drosophila
Fillip Port, Maja Starostecka, Michael Boutros
CRISPR-Cas12a-assisted PCR tagging of mammalian genes
Julia Fueller, Konrad Herbst, Matthias Meurer, Krisztina Gubicza, Bahtiyar Kurtulmus, Julia D. Knopf, Daniel Kirrmaier, Benjamin Buchmuller, Gislene Pereira, Marius K. Lemberg, Michael Knop
Amplification-free long read sequencing reveals unforeseen CRISPR-Cas9 off-target activity
Ida Höijer, Josefin Johansson, Sanna Gudmundsson, Chen-Shan Chin, Ignas Bunikis, Susana Häggqvist, Anastasia Emmanouilidou, Maria Wilbe, Marcel den Hoed, Marie-Louise Bondeson, Lars Feuk, Ulf Gyllensten, Adam Ameur
Instant FLIM enables 4D in vivo lifetime imaging of intact brains
Yide Zhang, Ian H. Guldner, Evan L. Nichols, David Benirschke, Cody J. Smith, Siyuan Zhang, Scott S. Howard
Customizable Live-Cell Imaging Chambers for Fluorescence and Super-Resolution Microscopy.
Adam L Tepperman, David Jiao Zheng, Maria Abou Taka, Angela Vrieze, Bryan Heit
Ultrastructural Visualization of 3D Chromatin Folding Using Serial Block-Face Scanning Electron Microscopy and In Situ Hybridization (3D-EMISH)
Paweł Trzaskoma, Błażej Ruszczycki, Byoungkoo Lee, Katarzyna K. Pels, Katarzyna Krawczyk, Grzegorz Bokota, Andrzej A. Szczepankiewicz, Jesse Aaron, Agnieszka Walczak, Małgorzata A. Śliwińska, Adriana Magalska, Michal Kadlof, Artur Wolny, Zofia Parteka, Sebastian Arabasz, Magdalena Kiss-Arabasz, Dariusz Plewczynski, Yijun Ruan, Grzegorz M. Wilczyński
Structure of the Lifeact–F-actin complex
Alexander Belyy, Felipe Merino, Oleg Sitsel, Stefan Raunser
A GT-seq panel for walleye (Sander vitreus) provides a generalized workflow for efficient development and implementation of amplicon panels in non-model organisms
Matthew L. Bootsma, Kristen M. Gruenthal, Garrett J. McKinney, Levi Simmons, Loren Miller, Greg G. Sass, Wesley A. Larson
SimpylCellCounter: An Automated Solution for Quantifying Cells in Brain Tissue
Aneesh Bal, Fidel Maureira, Amy A. Arguello
Adult mouse retina explants: an ex vivo window to explore central nervous system diseases
Julia Schaeffer, Celine Tardy, Floriane Albert, Stephane Belin, Homaira Nawabi
In Vivo Analysis of RNA Proximity Proteomes Using RiboPro
Xianzhi Lin, Kate Lawrenson
Fluorophore-labelled RNA aptamers to common protein tags as super-resolution imaging reagents
Juan Wang, Avtar Singh, Abdullah Ozer, Warren R. Zipfel
Research practice & education
Rethinking success, integrity, and culture in research (part 1) — A multi-actor qualitative study on success in science
Noemie Aubert Bonn, Wim Pinxten
Rethinking success, integrity, and culture in research (Part 2) — A multi-actor qualitative study on problems of science
Noemie Aubert Bonn, Wim Pinxten
A retrospective analysis of gender parity in scientific authorship in a biomedical research centre
Rinita Dam, Syed Ghulam Sarwar Shah, Maria Julia Milano, Laurel D Edmunds, Lorna R Henderson, Catherine R Hartley, Owen Coxall, Pavel V Ovseiko, Alastair M Buchan, Vasiliki Kiparoglou
The Ph.D. Panic: Examining the relationships among teaching anxiety, teaching self-efficacy, and coping in Biology graduate teaching assistants (GTAs)
Miranda M. Chen Musgrove, Elisabeth E. Schussler
Supervising the PhD: identifying common mismatches in expectations between candidate and supervisor to improve research training outcomes
Adam P.A. Cardilini, Alice Risely, Mark F. Richardson
Disadvantages of writing, reading, publishing and presenting scientific papers caused by the dominance of the English language in science: The case of Colombian Ph.D. in biological sciences
Valeria Ramirez-Castaneda
Survey of Australian STEMM Early Career Researchers: job insecurity and questionable research practices are major structural concerns
Katherine Christian, Carolyn Johnstone, Jo-ann Larkins, Wendy Wright, Michael R Doran
Study of the Influence of Preprint in bioRχiv for Peer Review and Acceptance Time of PLOS ONE
Hiroyuki Tsunoda, Yuan Sun, Masaki Nishizawa, Xiaomin Liu, Kou Amano
Development and validation of the Irish Science Self-Efficacy Children’s Questionnaire to assess the short-term influence of scientists facilitating outreach
Sarah Carroll, Jerome Sheahan, Veronica McCauley, Muriel Grenon
Methods for Running a Successful Women-in-STEM Organization on an Academic Campus
Deborah D. Rupert, Alexandra C. Nowlan, Oliver H. Tam, Molly Gale Hammell
| Why not…
Genetic Adaptation in New York City Rats
Arbel Harpak, Nandita Garud, Noah A. Rosenberg, Dmitri A. Petrov, Matthew Combs, Pleuni S. Pennings, Jason Munshi-South
 (No Ratings Yet)
(No Ratings Yet)
 The four letters of life – A, C, T and G – are ingrained into the scientific lexicon and burned into the brain of anyone who’s ever worked with or even just learned about genes, genomes and DNA. It’s a code that’s as inseparable from genetics as the double helix itself.
The four letters of life – A, C, T and G – are ingrained into the scientific lexicon and burned into the brain of anyone who’s ever worked with or even just learned about genes, genomes and DNA. It’s a code that’s as inseparable from genetics as the double helix itself.



 (5 votes)
(5 votes)