In vivo profiling of chromatin accessibility with CATaDa
Posted by Gabriel Aughey, on 13 March 2018
The following post is an introduction into the technnique described in our recent paper: Aughey, G.N., et al., CATaDa reveals global remodelling of chromatin accessibility during stem cell differentiation in vivo. Elife, 2018. 7.
Attempting to understand the biology of a complex organ, like the brain, comes with an array of technical challenges. Those of us who would like to understand development in the context of a living organism are faced with an enormously complex arrangement of cells, which may be superficially indistinguishable from one another, and furthermore, are inconveniently hidden away inside the body of an animal. Typically, if we want to examine the molecular characteristics of one of the constituent cell types of an organ, we need to physically isolate these cells from their biological context before applying the relevant assay. Cell separation can be very technically challenging and has been recently reported to cause artefacts in gene expression [1]. Therefore, methods in which cell types within a complex tissue can be examined without the need to remove them from their in vivo environment are of great interest to biologists working on in vivo models. Measuring the accessibility of chromatin to external factors is commonly used as a way of detecting functional regions of the genome. Techniques such as ATAC-seq or DNAse-seq are widely used for this purpose [2]. However, before now, we had no way of performing these chromatin accessibility assays without cell separation. In our recent paper we attempted to address this problem [3].
TaDa! – Chromatin interactions the easy way:
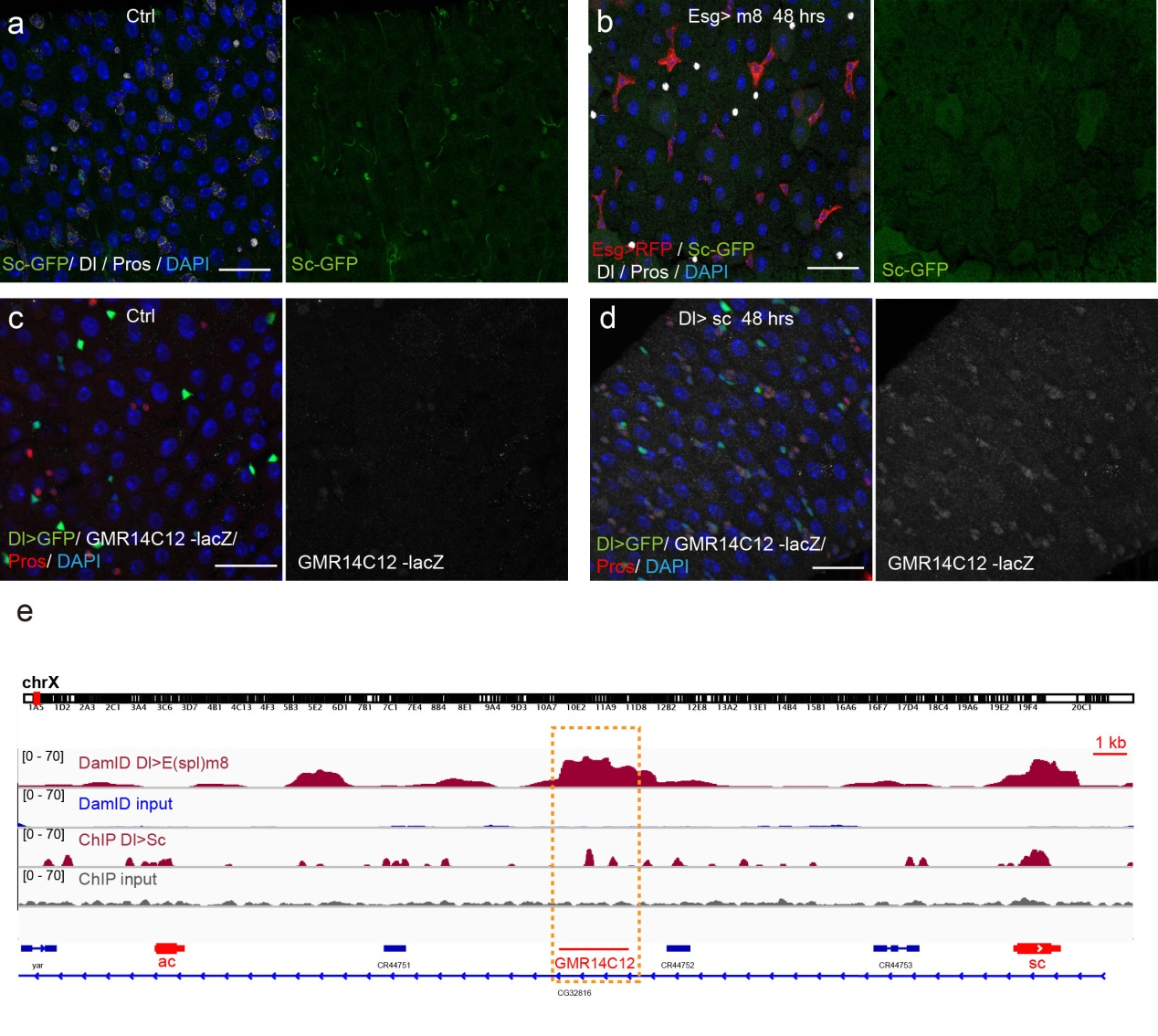
In the Southall lab we make extensive use of the Targeted DamID system (TaDa), which circumvents some of these issues when assaying protein-DNA interactions. This is a development of the well-established DamID system in which a chromatin-binding protein of interest is tethered to the E. coli adenine methylase, Dam [4]. Dam activity is relatively promiscuous, and so methylates any DNA that it comes into proximity with. Once this methylation is detected, we can infer the binding sites of a protein of interest. Targeted DamID expands on this approach by expressing the Dam-fusion protein as a bicistronic transcript, downstream of a primary open reading frame (ORF). This allows for very low level expression of the Dam-fusion so that the animal does not experience toxicity related to high Dam expression, and the methylation levels are not saturating – allowing for a semi-quantitative readout of protein binding [5]. Crucially, this construct can be combined with tissue specific binary expression systems (e.g. Gal4/UAS), allowing us to profile chromatin-protein interactions in a cell type of interest. (A full review of DamID and its applications can be found here: [6]).
Chromatin accessibility profiling with CATaDa
For TaDa experiments to work it is necessary to perform a control experiment in which untethered Dam protein is expressed in the cell-type of interest. The data produced from this control is then used to normalise the Dam-fusion binding data so that a reliable DNA-interaction profile can be generated. (This can be considered to have an analogous function to the input chromatin in a ChIP-seq experiment). Having generated much of this control data in the course of performing TaDa experiments, we wondered whether it might have applications beyond just normalisation. As Dam is highly active, we reasoned that the signal obtained after expression of untethered Dam would represent interactions between Dam and any regions of chromatin that it can access. Therefore, we inferred that Dam-only data may be a good proxy for chromatin accessibility.
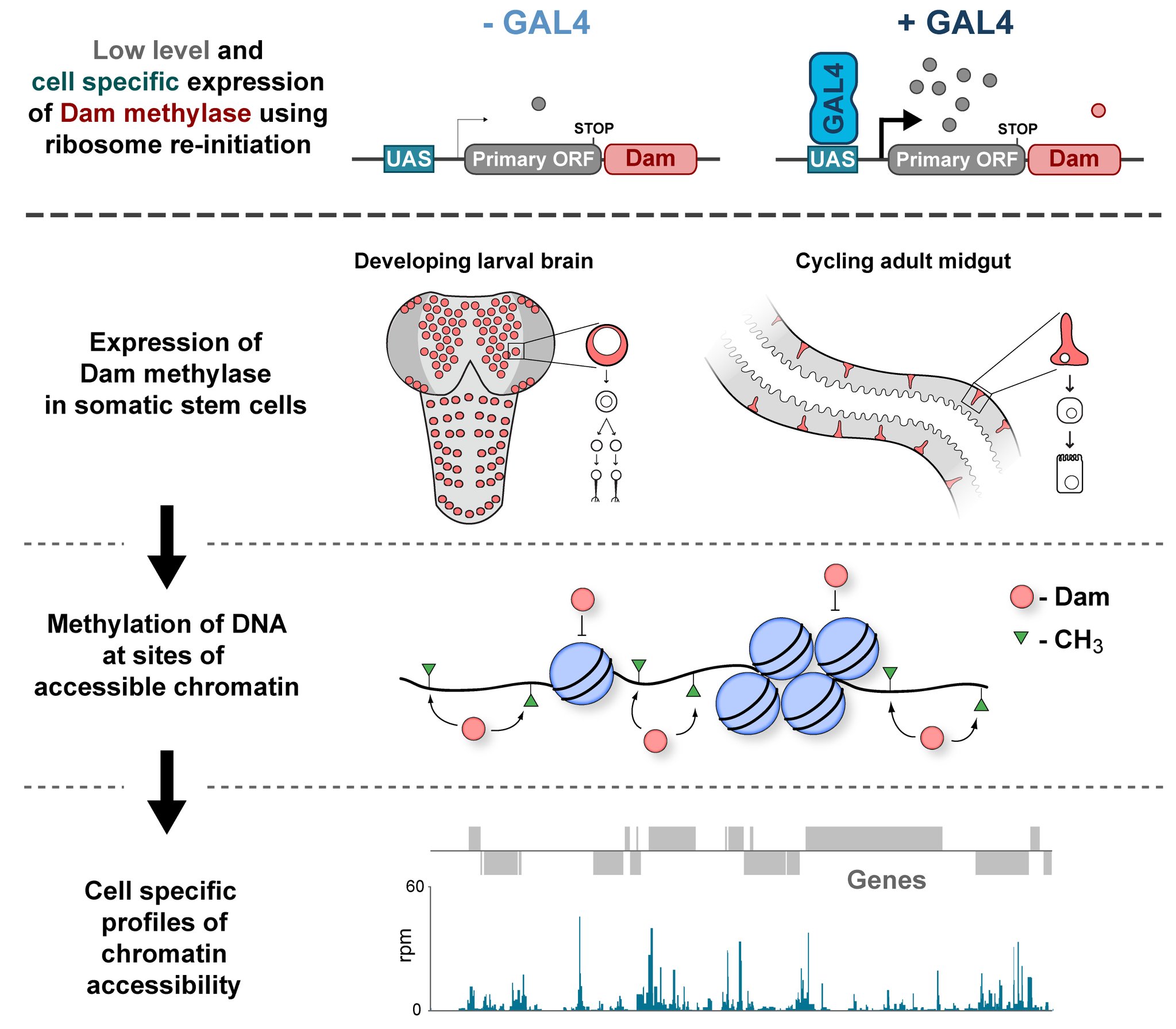
Given that we had gigabytes of Dam-only sequencing reads stored on our hard drives going unused, we were excited by the possibility that we could leverage these data into yielding further biological insights. To determine whether these data were comparable to established chromatin accessibility techniques, we compared some of our Dam data to previously published ATAC-seq or FAIRE-seq results [7]. We were excited to see that the signal from Dam methylation displayed many of the hallmarks of chromatin accessibility data that are observed with alternative techniques. For example, enriched signal at the transcription start site of genes. Overall, our data compared favourably to both ATAC and FAIRE-seq data. We found highly significant agreement between these datasets, and encouragingly, we saw that our data was comparable to ATAC-seq and out-performed FAIRE-seq in identifying previously validated enhancer regions. Therefore, we concluded that these data are appropriate for inferring meaningful chromatin accessibility data. We coined the term CATaDa (Chromatin Accessibility profiling using Targeted DamID) to describe this use of Dam methylation data for cell-type-specific profiling of chromatin accessibility.
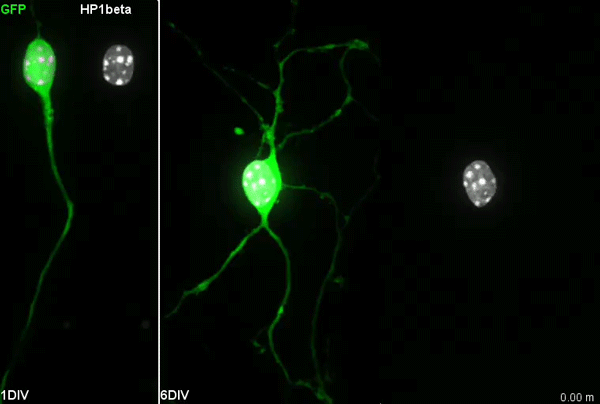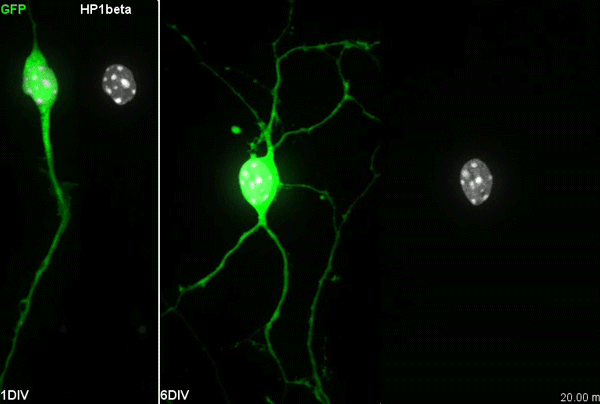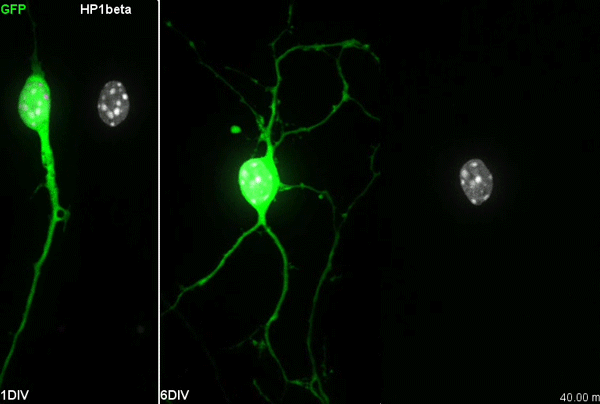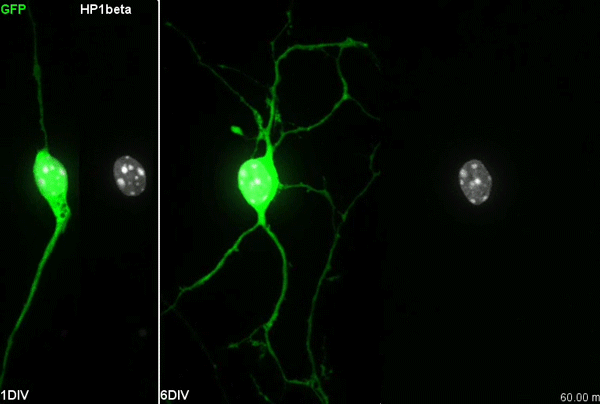
Chromatin accessibility during neurogenesis
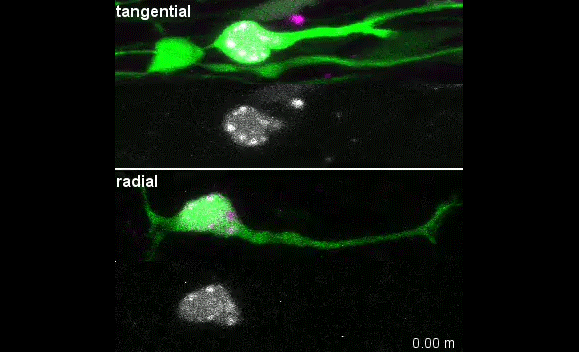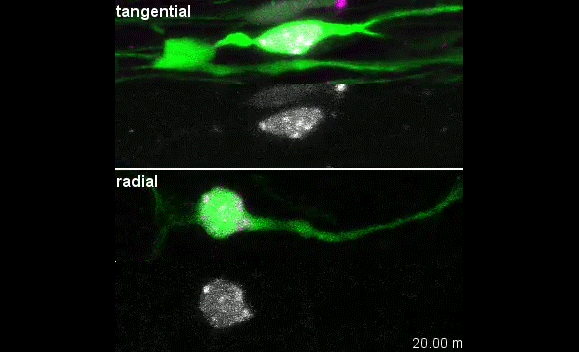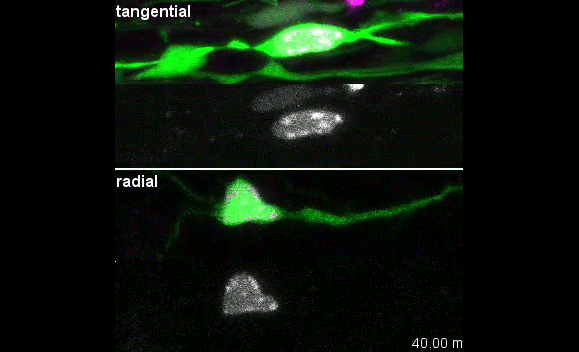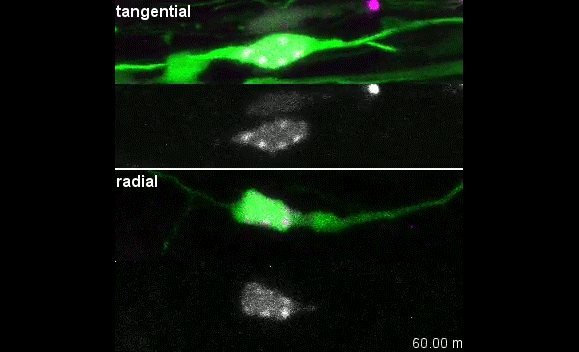
Our group is interested in understanding how the development of the nervous system is coordinated by changes to chromatin states. Therefore, we decided to apply CATaDa to the cells of the developing nervous system, including neural stem cells, their intermediate progeny (ganglion mother cells), and mature neurons. These data showed us that chromatin accessibility is incredibly dynamic between cell-types, with many regions becoming open or closed as development processes. Furthermore, we were able to examine regions of increased accessibility to detect enriched sequenced motifs which point towards factors that are involved specifically in various stages of nervous system development. We were also able to use our data to identify enhancers promoting cell-type specific gene expression.
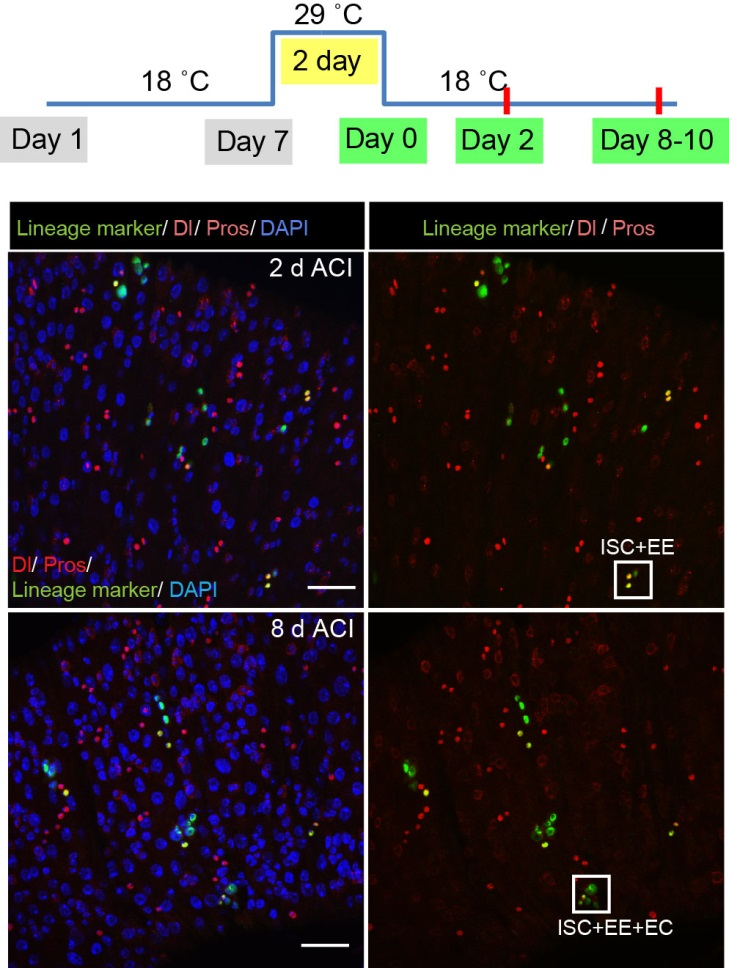
It is often said that chromatin is more open in stem cells, then becomes less accessible when differentiated. This has been shown to be the case when comparing embryonic stem cells to (e.g.) neurons [8]. This feature is often stated to be a general feature of stem cells, and is often applied to somatic stem cells (such as neural stem cells), despite the fact that we could find no evidence for the existence of any data attempting to examine this question in vivo. We realised that with our CATaDa data, we could answer this question by looking at the distribution of sequencing reads across the genome in each cell type. We were able to confirm that chromatin was globally more open in neural stem cells, which then becomes restricted in fully differentiated neurons. Interestingly, we saw that intermediate cell types showed intermediate chromatin accessibility which was closer to that of their stem cell progenitors. At individual loci this was also observed – regions of high accessibility associated with stem cell genes were also seen to be accessible (albeit at lower levels) in their progeny, despite the fact that expression of some of these genes is tightly restricted. To see if these trends were consistent in other tissues, we also assayed cells of the adult midgut. We chose this tissue because in contrast to neurogenesis, intestinal stem cells continue to divide throughout the lifetime of the animal for maintenance and repair. The previously described trends were also seen in these cells. Together these data suggest that chromatin accessibility decreases during the development of somatic lineages and intermediate cell types may have unexpected plasticity despite being fully committed to their cell fate.
Conclusion
We hope that we have shown that CATaDa is a useful approach for assaying chromatin accessibility that will appeal to researchers who are interested in in vivo approaches. Although we have only demonstrated CATaDA in flies, Targeted DamiD has recently been shown to be effective in mammalian cells, therefore our method can reasonably be expected to be effective in any model organism in which transgenic animals can be obtained.
Dam-ed if you do and Damned if you don’t – When to use CATaDa:
Yes:
- You want to profile chromatin accessibility of a subset of cells within a complex tissue / whole animal. (We typically start with dissected brains, but it is possible to use whole animals if the drivers used are specific enough)
- You have a limited amount of starting material (approx. 1000 cells minimum).
- You would like to profile the in vivo chromatin accessibility of a tissue, avoiding fixation or tissue dissociation artefacts.
- You have done a DamID/TaDa experiment with a conventional dam-only control and want to get the most out of your data!
No:
- You want to find the exact positioning of nucleosomes / enhancer sequences (CATaDa resolution is limited by availability of dam target motif – GATC).
- You need to detect changes in chromatin accessibility over very short timescales. Dam must be expressed over a relatively long time period for sufficient methylation to be detected. Consequently, the output of the experiment in slightly different to that of e.g. ATAC-seq (i.e. ATAC-seq represents a snapshot of a population of cells frozen in time, whereas CATaDa will include signal representing all accessible regions over the period assayed).
1. van den Brink, S.C., et al., Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat Methods, 2017. 14(10): p. 935-936.
2. Tsompana, M. and M.J. Buck, Chromatin accessibility: a window into the genome. Epigenetics Chromatin, 2014. 7(1): p. 33.
3. Aughey, G.N., et al., CATaDa reveals global remodelling of chromatin accessibility during stem cell differentiation in vivo. Elife, 2018. 7.
4. van Steensel, B. and S. Henikoff, Identification of in vivo DNA targets of chromatin proteins using tethered dam methyltransferase. Nat Biotechnol, 2000. 18(4): p. 424-8.
5. Southall, T.D., et al., Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: assaying RNA Pol II occupancy in neural stem cells. Dev Cell, 2013. 26(1): p. 101-12.
6. Aughey, G.N. and T.D. Southall, Dam it’s good! DamID profiling of protein-DNA interactions. Wiley Interdiscip Rev Dev Biol, 2016. 5(1): p. 25-37.
7. Davie, K., et al., Discovery of transcription factors and regulatory regions driving in vivo tumor development by ATAC-seq and FAIRE-seq open chromatin profiling. PLoS Genet, 2015. 11(2): p. e1004994.
8. Meshorer, E. and T. Misteli, Chromatin in pluripotent embryonic stem cells and differentiation. Nat Rev Mol Cell Biol, 2006. 7(7): p. 540-6.
 (No Ratings Yet)
(No Ratings Yet)




 (5 votes)
(5 votes)

 (8 votes)
(8 votes)