The BFSCI serves as a hub for basic and translational research in stem cell biology and regenerative medicine across the medical school campus and is located in state-of-the-art research space.
The CDRB Department ranks as one of the top cell/developmental biology departments in the country, with a focus on cell biology of developmental proesses and epithelial organ development, utilizing multiple model systems to delve into fundamental questions of biology and disease pathogenesis.
We seek highly interactive and collaborative investigators whose research interests will complement and enhance those of the Department and/or the Institute.
Faculty positions in CDRB: Assistant or Associate Professor focusing on vertebrate niche stem cell biology, cell biology, epithelial biology, and/or developmental biology
Faculty positions in BFSCI: Assistant or Associate Professor focusing on basic or translational mammalian stem cell biology, tissue homeostasis and/or regenerative medicine
Candidates should hold a Ph.D., M.D., V.M.D., M.D./Ph.D. or equivalent degree and are expected to participate in research and graduate student mentoring and teaching. The Icahn School of Medicine provides an exceptional intellectual environment with outstanding facilities; generous start-up packages are available.
Applicants should submit a single PDF file containing a cover letter, CV, and a 2-3 page statement of research interests. Please name the file as: Applicant’sLastName.Applicant’sFirstName.pdf.
Applicants should also arrange to have three individuals send confidential letters of reference. Please have the letters named as: Applicant’sLastName.Applicant’sFirstName_Referee’sLastName.pdf.
Please indicate in your cover letter whether you are applying for a position in CDRB, BFSCI, or both.
Completed applications and letters of reference should be submitted via email to Taylor Stokelin: taylor.stokelin@mssm.edu and will be reviewed on a rolling basis with an initial deadline of November 1, 2020.
The Icahn School of Medicine at Mount Sinai is an Equal Opportunity and Affirmative Action Educational Institution and Employer. Women and members of underrepresented minority groups are strongly encouraged to apply.
We seek a Lab Manager. Our laboratory uses genomics, organoids and mouse models to study mammalian kidney in health and disease. We possess substantial expertise in single cell omic and multi-omic approaches including all aspects of informatic analysis. Examples of our recent work can be found here:
Our supportive environment values team work. We seek a lab manager to oversee and coordinate lab functioning and compliance but also to design, conduct and oversee research projects. We are located in recently renovated space in the McDonnell Medical Science Building. The successful applicant will have substantial lab experience and writing skills. This position offers superb opportunities for continued scientific learning in a well-funded laboratory, and continued career development including authorship, and is full time. Salary range $51 – $91k.
The successful applicant will hold the title of Lab Manager. The Humphreys Lab is a collaborative, highly productive and supportive scientific work environment.
Humphreys Laboratory:
The Humphreys lab consists of eight post-doctoral fellows, three graduate students and a lab manager. An overview of the Humphreys Lab research can be found at: http://humphreyslab.com/ and on Twitter @HumphreysLab
To apply for this position, please submit a letter explaining your interest, a CV and contact information for references to Benjamin Humphreys MD, PhD (humphreysbd@wustl.edu)
Please, always run specificity controls with Morpholinos. Morpholinos can reveal gene functions that can be concealed by genetic compensation in a mutant, but rigorous specificity controls are essential to determining whether a morphant phenotype is due to knockdown of the oligo target or an unexpected interaction with a different RNA. The Morpholino-in-a-mutant approach described in Jiang et al. 2020 (linked below) is an excellent approach when a mutant is available.
For those using Morpholinos, these three papers provide important perspective on the need for and good design of specificity controls.
On the mechanism behind many differing phenotypes of morphants and mutants:
Sztal TE, Stainier DYR. Transcriptional adaptation: a mechanism underlying genetic robustness. Development. 2020 Aug 14;147(15):dev186452. doi: 10.1242/dev.186452.
https://dev.biologists.org/content/147/15/dev186452
Describing a case where the mutant – morphant phenotypic difference is not caused by transcriptional adaptation:
Jiang Z, Carlantoni C, Allanki S, Ebersberger I, Stainier DYR. Tek/Tie2 is not required for cardiovascular development in zebrafish. Development. 2020 Sep 14:dev.193029. doi: 10.1242/dev.193029.
https://dev.biologists.org/content/early/2020/09/12/dev.193029
On Morpholino controls:
Stainier DYR, Raz E, Lawson ND, Ekker SC, Burdine RD, Eisen JS, Ingham PW, Schulte-Merker S, Yelon D, Weinstein BM, Mullins MC, Wilson SW, Ramakrishnan L, Amacher SL, Neuhauss SCF, Meng A, Mochizuki N, Panula P, Moens CB. Guidelines for morpholino use in zebrafish. PLoS Genet. 2017 Oct 19;13(10):e1007000. doi: 10.1371/journal.pgen.1007000. eCollection 2017 Oct.
http://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1007000
Our body consists of a multitude of highly specialized tissues: the neurons in our retina seem to have little in common with the glandular cells in the intestine or our muscle fibers. Yet nothing hints at that complexity at the time of conception, when the genomes of sperm and egg fuse to define the blueprint of a new individual. Indeed, how an entire organism arises from a single cell truly is one of the most fascinating questions of biology. As the embryo develops, its building blocks (cells) gradually assume specialized functions and coordinately go through transient states that serve as intermediate steps wherefrom more complex structures will be molded as time progresses. Throughout this process, cells dynamically receive and send information to instruct their neighbors to assume specific fates. This cell-to-cell communication ensures all parts working together perfectly to create a self-organizing system that will eventually give rise to an entire body. Embryonic development thus follows a delicate choreography wherein every cell has to fulfill a precise role depending on its relative position in space and time. Unraveling the blueprint of the making of an embryo is a daunting endeavor that has fascinated developmental biologists for decades.
This study started with two physicists-turned-biologists (Pierre Neveu and Lars Hufnagel) marveling at SPIM (selective plane illumination microscopy) movies of developing Phallusia mammillata embryos, an ascidian species that is an ideal model for advanced imaging approaches due to the high optical transparency of early embryos. Our ambition was ignited to take a quantitative systems approach to decipher the molecular mechanisms underpinning the marvelous unfolding of embryonic complexity captured in those movies. We set out to employ high resolution single-cell RNA sequencing (scRNA-Seq) to decipher the complete transcriptome of every single cell in the developing embryo from zygote to gastrulation.
We quickly came to realize that the choice of an appropriate model system would be critical for the success of this highly ambitious project. Neither of us had previously worked on ascidian development but we embraced Phallusia mammillata for its many advantageous properties. Ascidian embryos are made up of a very small number of cells and their development is stereotypical. This means that the developmental history of any given cell is known but it also means that its future fate can be predicted and all individuals develop according to that same invariable blueprint. However, ascidians have a genetic makeup that resembles the ones of vertebrates (albeit less complex as genome duplication has not occurred).
We thought that possessing the complete (or near complete) collection of cells of individual embryos would enable us to ask two fundamental questions (Figure 1): could one actually determine the physical position of a given cell from its gene expression? How variable is gene expression during development?
Figure 1. A: Both the lineage history of a cell and its spatial position will influence its gene expression. B: The bilateral symmetry of Phallusia mammillata embryos allows to compare the reproducibility of gene expression in bilaterally symmetric cell pairs within one embryo and across embryos.
The small number of cells that make up a Phallusia embryo at gastrulation brings a crucial advantage. It allowed us to sample a large number of individuals at moderate costs in order to compare how different the gene expression is between individual embryos of the same stage.
Moreover, ascidian embryos are bilaterally symmetric so in theory there should be two copies of every cell in a given embryo (one for the left part and one for the right part of the embryo). This solves a fundamental conundrum of any single-cell approach: How to get hold of a suitable biological replicate?
Cell type identification in individual embryos
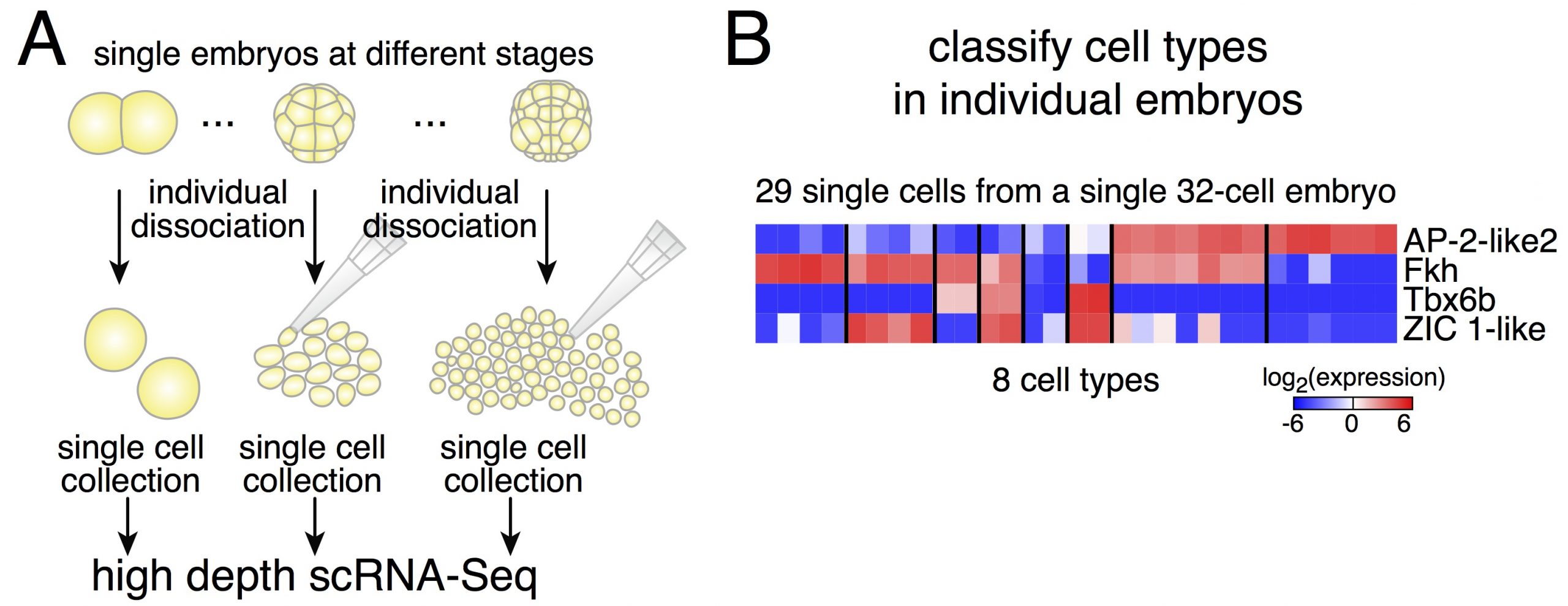
Dissociating single embryos up to gastrulation, we performed RNA-Seq at very high depth. However, a first obstacle we had to tackle was the lack of a published annotated transcriptome for Phallusia. After performing the de novo assembly of the Phallusia transcriptome, it turned out that we could capture quantitatively in single cells the expression levels of as many genes as for bulk mRNA-Seq on pools of embryos.
Figure 2. A. Strategy to capture expression profiles of cells from individual embryos. B: The expression of only four genes is sufficient to classify cells into the 8 cell types present in 32-cell embryos.
Due to potential variability between embryos, we thought it might be simpler to classify cells within a given embryo (and then compare cell types across embryos of the same stage, Figure 2). Relying on an iterative classification scheme, we identified 18 cell types in a 64-cell embryo. This diversity is striking despite the small number of cells. Another interesting point is the low dimensionality of the classification: the number of gene groups that can classify cell types is smaller than the number of cell types. It means that cell fate specification relies on the modular expression of genes (and not only on genes that would be exclusively expressed in individual cell types as what would be commonly assumed).
Reconstructing lineage trajectories from scRNA-Seq data
Equipped with the exact cell type composition of the embryo after each round of cell divisions, we then asked what is the mother-daughter relationship between cell types. When matching cell types across stages, correct accounting must factor in cell divisions, in other words there should be twice as many cells in the daughter cell types of a given mother cell type. Indeed, if one does not keep track of cell divisions, one would fail to capture offshoots of the germ cell lineage (which is itself transcriptionally silent up to the 64-cell stage and therefore stands out compared to other cell lineages).
Two findings stand out from the lineage reconstruction. First, we only found asymmetric mRNA inheritance between daughter cells in the germ cell lineage. Our dataset thus dispels the notion of mosaic development (at least relying on RNA-based factors) for all the somatic lineages. Second, specification is driven by three successive waves of upregulation of transcription factors. This stepwise cascading principle of transcription factors was valid for all somatic lineages.
Reconstructing the spatial organization of the embryo from scRNA-Seq data
Development is a dynamical process in which time and space are fundamentally linked. The gene expression of a given cell will adapt to the signals it receives from its surroundings in a history-dependent manner. Reversing the above statement, the spatial and temporal identity of every cell should be encoded in its gene expression signature. This is a crucial point as we had not kept track of the spatial origin of cells during the dissociation of the embryos. Thus, the next logical step was to see if we could reconstruct the physical organization of the embryo directly from the expression profiles of all its cells.
A perfect starting point is when the embryo activates its genome. Cells are a blank slate (except the ones that inherit maternal determinants, the future germ cells in the case of Phallusia) and will respond to the morphogenetic cues present in their environment. Expression profiles of the cells of 16-cell embryos define two axes: one separates the two embryo poles and the other one corresponds to the anteroposterior axis. Remarkably, the expression profiles of the different cells project in a way that mirrors the cells’ physical positions in the embryo. It thus appears that the future germ cells (through the inheritance of maternal determinants, among which a Wnt ligand) act as a signaling center that organizes the initial patterning of the embryo at 16-cell stage. The same map recapitulates the relative positions of the 8 cell types present in 32-cell embryos but can no longer resolve a few sister cell types at the 64-cell stage. In fact, different signaling ligands are upregulated at the 16- and 32-cell stages, changing the morphogenetic cues cells experience. The introduction of new dimensionality reduction axes at the 64-cell stage is thus necessary and theses axes map faithfully the position of the different cell types of the vegetal pole. In practice, one axis corresponds to the anteroposterior axis and the other to the medial-lateral axis.
Hence, the spatial organization of the embryo is reflected in the gene expression of individual cells and it can be reconstructed from it. Cells read out the local signaling environment and adapt their gene expression accordingly.
After assembling the Phallusia transcriptome, it became apparent that Phallusia possesses orthologs for all the transcription factors and signaling molecules expressed during embryogenesis annotated in the Ciona genome. There is a remarkable conservation of the expression territories of ortholog genes between the two species. Thus, there is a strong regulative dimension to ascidian development despite the limited number of cells constituting their embryos.
Assessing the variability and reproducibility of gene expression during development
Our dataset has both high cell coverage of individual embryos but also many embryos of a given stage. We could then study the reproducibility of gene expression within an embryo and across embryos.
How similar are the two sides of the embryo? Here again, the small number of cells in the embryo allows an unambiguous comparison. Some cell types have only two cells per embryo so there is necessarily one coming from the left side of the embryo and one from the right side. In the majority of these cell pairs originating from the same embryo, the expression of marker genes is highly reproducible with a precision comparable to the one of housekeeping genes.
While bilaterally symmetric cells in a given embryo are highly similar, there is an obvious spread in gene expression profiles within particular cell types. What could account for these differences? The most natural explanation would be that they are due to slight differences in staging. In other words, cells with lower expression levels are younger than cells with higher expression levels. An alternative explanation would be that gene expression is noisy and cells have different expression levels that do not influence normal development. However, an analysis called RNA velocity allows to extrapolate the future expression levels of a given gene by looking at the ratio between spliced and unspliced counts. RNA velocity supports the notion that cells are caught at different times along the upregulation trajectory of these genes. The coordinated upregulation of dozens of genes in a given cell type is in fact accurately modeled with a model of linear variation in time. Thus, expression differences between embryos and between cells of the same cell type can be attributed to slight differences in developmental timing. It appears that transcriptional programs guiding cell fate unfold according to a temporally well-choreographed sequence in a given cell type.
Projecting the single cell expression profiles onto a fully segmented movie of embryonic development enabled us to generate a digital embryo, i.e. a 4D representation of embryonic development accounting for every cell and every gene. This data can be explored and visualized at digitalembryo.org, where anyone can generate their own movies of the temporal and spatial gene expression dynamics oftheir genes of interest.
Charting an atlas of a developing embryo at the single-cell and whole genome level would have seemed an impossible mission a mere decade ago. The merge of developmental biology and quantitative approaches from systems biology holds great promise for the future. While traditional studies have focused on the role of individual genes in specific lineages, unraveling the complexity of the making of an entire organism will greatly benefit from the holistic perspective made available by the constant advance of single-cell techniques.
The Rohner Lab at the Stowers Institute for Medical Research has an opening for a Research Specialist to further develop tools for the emerging research organism Astyanax mexicanus. Visit http://research.stowers.org/rohnerlab/ for more information on the research in the lab.
The selected candidate will help with day to day operations of the lab and develop new tools for the cavefish system, such as transplantions, genome editing, viral mediated gene transfer, generation of cell lines, transgenic lines and others. The candidate will receive strong support from the core facilities that provide advice, training and service to enhance the Institute’s interdisciplinary and collaborative research programs. Current core facilities are staffed by over 100 scientists with expertise in bioinformatics, cytometry, histology, imaging, microarray, next generation sequencing, transgenic and ES cell technologies, proteomics and molecular biology. The Stowers Institute offers a highly competitive compensation and benefits package.
The Rohner Lab has a strong commitment for mutual success and is dedicated to providing support for all lab members. Minimum requirements include a doctoral degree in the life sciences, chemistry, or biomedical engineering with significant postdoctoral experience in one or more of the following areas: molecular biology, developmental biology, genetics, genomics, evodevo, physiology.
In addition to excellent verbal and written communication skills, successful candidates must be dynamic and able to motivate others, and being creative and proficient at problem solving.
Application Instructions: To apply, please submit (1) a brief cover letter, (2) a current CV, and (3) contact information for two professional references to Dr. Nicolas Rohner at nro@stowers.org cc: careers@stowers.org. Applications due on October 15th, 2020, after that position will remain open until filled.
The Rohner Lab at the Stowers Institute for Medical Research has an opening for a Postdoctoral Researcher to develop an independent project investigating the molecular, genetic, or developmental mechanisms of how cavefish thrive under extreme metabolic conditions. The lab has previously found that the cavefish Astyanax mexicanus develop high-blood sugar and insulin resistance as part of their natural strategy to survive in the caves but without the usually associated health problems (Riddle et al. Nature. 2018 Mar 29;555(7698):647-651). Furthermore, we recently found that cavefish are resilient to developing inflammation in adipose tissue due to a switch from innate to adaptive immunity (Peuss et al. Nature Ecology & Evolution 2020 Jul 20). Visit http://research.stowers.org/rohnerlab/ for more information.
The selected candidate will investigate the molecular mechanism underlying these or other impressive adaptations. The candidate will closely work with the core facilities at the institute to perform single-cell RNA sequencing, proteomics, and functional validation in vitro and in vivo. A computational/bioinformatic focus is also possible. The candidate will receive strong support from the core facilities that provide advice, training and service to enhance the Institute’s interdisciplinary and collaborative research programs. Current core facilities are staffed by over 100 scientists with expertise in bioinformatics, cytometry, histology, imaging, microarray, next generation sequencing, transgenic and ES cell technologies, proteomics and molecular biology. The Stowers Institute offers a highly competitive compensation and benefits package.
The position is funded for an initial period of one year but can be renewed for up to five years in order to allow enough time to develop a research program/publication record that makes the postdoc a strong candidate for an independent position. The Rohner Lab has a strong commitment for mutual success and is dedicated to providing support for all lab members.
Minimum requirements include a doctoral degree in the life sciences, chemistry, or biomedical engineering. Experience in one or more of the following areas is desirable: molecular biology, developmental biology, genetics, genomics, evodevo, physiology.
In addition to excellent verbal and written communication skills, successful candidates must be dynamic and highly motivated, work independently and creatively, able to work in a team-oriented environment, and proficient at problem solving.
Application Instructions: To apply, please submit (1) a brief cover letter, (2) a current CV, and (3) contact information for two professional references to Dr. Nicolas Rohner at nro@stowers.org cc: careers@stowers.org. Applications due on October 15th, 2020, after that position will remain open until filled.
Winged insects are the most diverse and numerous group of animals on Earth. This great diversity has been possible thanks to the acquisition of novel morphologies and lifestyles. How the changes in their genomes contributed to the appearance and evolution of these traits is key to understand how this lineage adapted and conquered the huge plethora of ecosystems that they inhabit nowadays.
C. dipterum male individual
Mayflies (Ephemeroptera), together with dragonflies and damselflies (Odonata) form the Paleoptera group, which is the sister group of the rest of winged insect lineages. Extinct Paleoptera are thought to be the first insects that developed wings and a partial metamorphosis: juveniles have to undergo physiological and morphological changes to get into their final adult forms. In the case of extant Paleoptera, these changes are quite striking, as juveniles (nymphs) are aquatic until they emerge from the water to become flying adults. In addition, the Baetidae family of mayflies have evolved a remarkable sexual dimorphism. Males have an extra pair of eyes located in the dorsal part of the head called the turbanate eyes due to their turban-like shape. All these features make mayflies a key group of insects to investigate the genomic adaptations to different ecological niches, how visual systems are specialized and how wings appeared in the first insects during the Carboniferous period.
Eggs about to hatch
We recently sequenced the genome of the mayfly Cloeon dipterum (Almudi et al., 2020) and looked for signatures of adaptations to different lifestyles and the origin of insect wings within it. Together with the genomic DNA, we also profiled gene expression across multiple tissues, organs and developmental stages (a total of 37 different samples). This publication is the culmination of a scientific endeavour that started five years ago in the Casares lab, in Seville, when I was granted a Marie Sklodowska-Curie fellowship and established C. dipterum as an emergent model system (see https://thenode.biologists.com/day-mayfly-lab/lablife/and Almudi et al., 2019).
The sequencing of the genome and the transcriptomes revealed many interesting aspects relevant not only for the Developmental and Evolutionary Biology fields but also for Physiology, Ecology or conservation. We discovered striking expansions of sensory gene families such as the Odorant Binding Proteins (with described chemoreception function) and the UV and Blue light-sensitive Opsins (which are the main molecules sensing light).
One of our most significant results is the deep conservation of gene expression across tissues in Arthropods. Our RNA-seq datasets allowed us to generate mayfly gene clusters based on gene expression across our multiple samples and to make pairwise comparisons of these clusters to the ones also generated for the fruitfly Drosophila melanogaster and the centipede Strigamia maritima, a non-insect arthropod. To our surprise, we found many orthologous genes that were expressed in wings, of both, the mayfly and the fruitfly, indicating transcriptomic conservation. Moreover, when we tested the function of some of these genes with previously unknown function, we found that all of them participated in wing development. Thus, our transcriptomic comparative approximation comes out as a very helpful approach to infer function of new genes. Interestingly, using the same analysis, we identified a set of genes shared by the nervous system of flies, mayflies and centipedes.
A gill plate showing specific staining against a chemosensory gene (OBP219) and HRP (a neural marker)
Finally, we also wanted to contribute to solving one of the long-standing questions in evo-devo: how did insect wings originate? Although some advances have been made in the recent years, it is still an open issue how wings first appeared in the first winged insects and which were the genes and the structures that gave rise to them. Two features make mayflies ideal to investigate this question: First, as it was mentioned earlier, their ancestors were the first insects that had the ability of flying. And second, the aquatic larvae (nymphs) of fossil and extant mayflies possess abdominal beating gill plates, which have been suggested to be serial homolog structures to the thoracic wings. Thus, we could not miss the opportunity to have a first transcriptomic approximation to the problem. We found that the gill plates transcriptome is the most similar to the wing transcriptome within our dataset and that when a gene is specifically expressed in wings and a second tissue (out of our samples), this second tissue tend to be the gill plate in most cases, suggesting a close relation between these two organs. Of course, more experiments are necessary to answer whether gills and wings share a common origin, but our research on mayflies opens the possibility of addressing the ‘origin of insect wings’ problem from a new perspective and using a key organism, a mayfly, due to its position in the phylogeny of insects.
I hope that the establishment of C. dipterum in the laboratory and the sequencing of its genome set the foundation of multiple research lines to be pursued by us and others to unveil the many wonders of winged insects.
Note: This project has been possible thanks to the joint efforts of all my amazing co-authors: Carlos Martín-Blanco, Isabel García-Fernández, Adrián López-Catalina, Joel Vizueta, Chris Wyatt, Alex de Mendoza, Ferdinand Marlétaz, Panos Firbas, Roberto Feuda, Giulio Masiero, Patricia Medina, Ana Alcaina, Fernando Cruz, Jessica Gomez-Garrido, Marta Gut, Tyler S Alioto, Carlos Vargas-Chavez, Kristofer Davie, Bernhard Misof, Josefa González, Stein Aerts, Ryan Lister, Jordi Paps, Julio Rozas, Alejandro Sánchez-Gracia, Manuel Irimia, Ignacio Maeso and Fernando Casares.
Almudi, I., Vizueta, J., Wyatt, C.D.R., de Mendoza, A., Marletaz, F., Firbas, P.N., Feuda, R., Masiero, G., Medina, P., Alcaina-Caro, A., Cruz, F., Gomez-Garrido, J., Gut, M., Alioto, T.S., Vargas-Chavez, C., Davie, K., Misof, B., Gonzalez, J., Aerts, S., Lister, R., Paps, J., Rozas, J., Sanchez-Gracia, A., Irimia, M., Maeso, I., Casares, F., 2020. Genomic adaptations to aquatic and aerial life in mayflies and the origin of insect wings. Nat Commun 11, 2631.
We are looking for a highly motivated Senior Laboratory Research Scientist to join the Santos laboratory headed by Dr Silvia Santos at the Francis Crick Institute. The lab focuses on understanding cell decision-making. Current areas of research include understanding regulatory mechanism of cell division and cellular differentiation, using human embryonic stem cells as a model system. There is a strong focus on single cell analysis, live cell imaging and genomics. The team is currently composed of two PhD students, one undergraduate student and three post-doctoral fellows.
For more information please see https://www.crick.ac.uk/research/labs/silvia-santos
Who are you?
The successful post holder is keen to drive hers/his own research, support on-going collaborative research and help with lab management and/or training. The ideal candidate is likely to be an energetic, organised individual who thrives to work on interesting biological problems in a highly collegial and collaborative work environment.
Excellent people and organisation skills are essential.
Why is the post awesome
This post provides the successful candidate with career progression opportunities, potential for a permanent research job, mentorship and a fantastic wider support network at the Crick Institute.
We are looking for a highly motivated Senior Laboratory Research Scientist to join the Santos laboratory headed by Dr Silvia Santos at the Francis Crick Institute. The lab focuses on understanding cell decision-making. Current areas of research include understanding regulatory mechanism of cell division and cellular differentiation, using human embryonic stem cells as a model system. There is a strong focus on single cell analysis, live cell imaging and genomics. The team is currently composed of two PhD students, one undergraduate student and three post-doctoral fellows.
For more information please see https://www.crick.ac.uk/research/labs/silvia-santos
Who are you?
The successful post holder is expected to drive hers/his own research, support on-going collaborative research and help with lab management and/or training. The ideal candidate is likely to be an energetic, organised individual who thrives to work on interesting biological problems in a highly collegial and collaborative work environment.
Excellent people and organisation skills are essential.
Why is it awesome
This post provides the successful candidate with career progression opportunities, potential for a permanent contract, mentorship and a fantastic wider support network at the Crick Institute.
 (No Ratings Yet)
(No Ratings Yet) (1 votes)
(1 votes)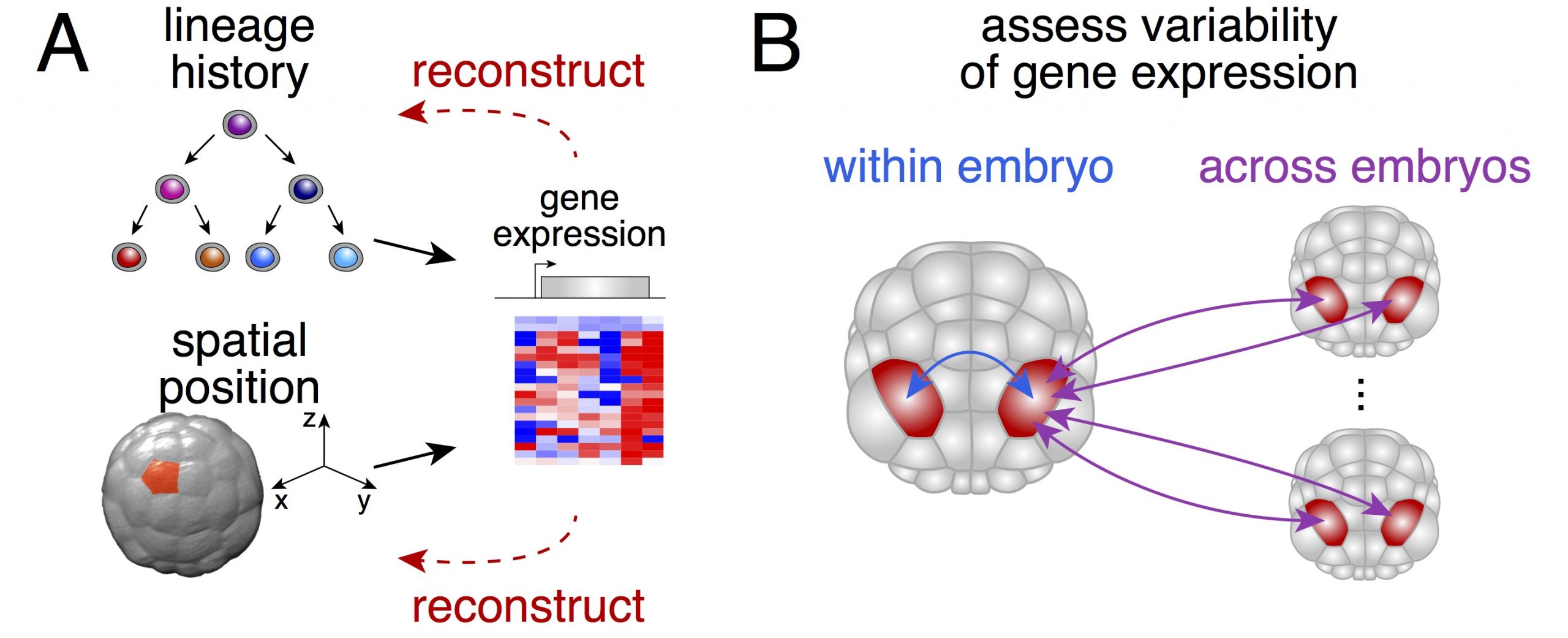
 (11 votes)
(11 votes)