Department of Physiology, Anatomy and Genetics, Oxford
Grade 7: Salary in the range £31,076 – £32,004 p.a.
Applications are invited for a Postdoctoral Research Scientist to join Professor Mathilda Mommersteeg’s laboratory to work on a project focussed on identifying the mechanisms underlying fish heart regeneration using the Mexican cavefish, Astyanax mexicanus. The post would be ideally suited to a postdoctoral scientist with experience in working with fish models and background expertise in heart development or regeneration.
The successful applicant will be specifically charged with performing QTL analyses to link the ability for heart regeneration to the genome, followed by functional validation of the identified loci. The main techniques in this project will include fish cardiac injury surgery, the generation of constructs for injection to generate transgenic zebrafish and Astyanax mexicanus lines using CRISPR, oocyte injections, western blotting, qRT-PCR, immunohistochemistry and in situ hybridisation.
You must hold, or be near completion of a PhD (or equivalent) in molecular biology, biomedical sciences, genetics or a related science and have experience of working with in vivo models of development and/or cardiovascular disease.
You will be based in the Sherrington Building in the Department of Physiology, Anatomy and Genetics at the University of Oxford.
The position is funded by an ERC starting grant for up to 5 years. The post is due to start on 1 March 2017.
The closing date for applications is 12.00 noon on Monday 19 December 2016. Interviews are likely to be held early January 2017.
“It’s fair to say that we currently know more about the moon than about our own embryonic development. The current textbooks all show the same kind of images based on a few embryonic specimens from the 1930s. Some of those are not even human embryos. New version of these books keep using those images, often without reference, only updating new molecular information”
This leads to the situation that the origin of the knowledge on human embryology became forgotten and that some of the knowledge was based on chicken, axolotl and mouse specimens. Moreover, the complex morphological changes during development cannot be well illustrated on the flat two-dimensional pages of text books.
To remedy this situation we proposed to use the sections of the embryos available in the Carnegie Collection to generate a comprehensive series of interactive 3D reconstructions of human embryonic development; a proposal that was based on the availability of 3D reconstruction and visualisation software.
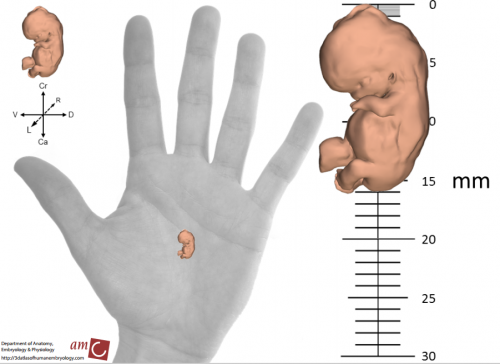
3D view of a reconstructed stage 20 human embryo, from Figure 2, de Bakker, et al. Science, 2016
This project, initiated by Professor Antoon Moorman, with dedicated financial support of the Academic Medical Center in Amsterdam, the Netherlands, was carried out by Bernadette de Bakker. The educational angle made that the project was ‘by students – for students’. De Bakker obtained a duplicate series of images of stages 7 to 23 (15 – 60 days of development) from the Carnegie Collection in Washington DC. After alignment in a 3D reconstruction program, a total of 75 medical students manually annotated every structure and organ in each of the about 15,000 images as part of their scientific internship; an effort that took more than 45,000 hours.
Another 15 students, this time from the game development faculty, then ‘modelled’ the resulting 3D reconstructions to reduce the complexity without losing biological detail. Their results, turned into 3D-PDF files, thus represent the actual morphology of the series of embryos in a format that can be easily interactively handled with the commonly used PDF reader. This was all supervised and supported by the expert embryologists of the department.
“The 3D Atlas and Database of Human Embryology is the first to present such a large amount of data based on such a large number of human embryos. With this atlas and database we are able to quantify very precisely embryonic growth and the growth and position of specific organs”
Stage 17 embryo, scaled, from the interactive PDF available on the 3D atlas website.
The 3D atlas, that contains morphological reconstructions of 14 stages, is accompanied by a database with quantitative data based on 17 duplicate stages of reconstructed embryos. This database shows the total volume of the embryo grows exponentially with a constant volume increase of 25% per day. However, different organs grow with different growth rates, depending on their function during development. The project team established a tool to determine the position of organs relative to the developing vertebral column of the embryo. Application of this tool can help to solve ambiguities with respect to the relation between organs. The third series of data relates the chronology of human development to that of the mouse and the chicken. These data show that each species runs its own embryological script.
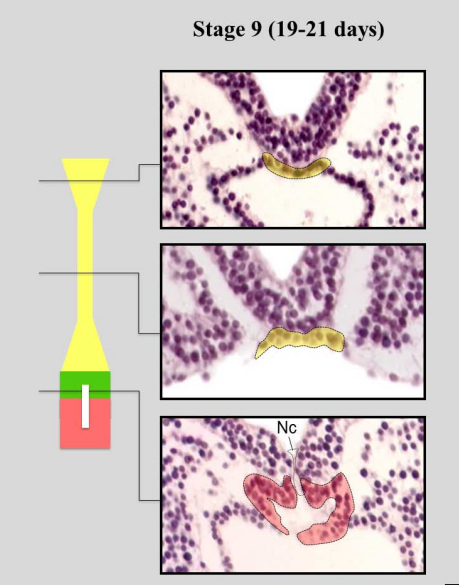
Notochord development at Stage 9, from Figure 6, de Bakker, et al. Science, 2016.
Already during the construction of the atlas, discrepancies between the observed embryology and the embryology textbooks became apparent. The textbook descriptions of the development of the venous system and the notochord clearly show influence of knowledge dissipating from studies on lower vertebrates. Descriptions that could not be corroborated in the current reconstructions based on the real human substrate. The interactive 3D atlas, and the source images that are also available from the website, allow every user, be it student or biomedical specialist, to independently verify those discrepancies and form their own opinion.
“The atlas is useful because to study how congenital malformations appear it is important to know how normal human embryonic development occurs. Experimental embryologists will know how the changes in their experimental animals relate to the human situation. Scientists who study how toxic substances can affect embryonic development, are helped to refine the chick and mice models they work with. Moreover, also gynaecologists can use this atlas to show pregnant women how their child develops.”
Contributors: Bernadette de Bakker, Jan M Ruijter and Antoon Moorman
The 3D atlas of human embryology is available here:
We are delighted to invite you to our one-day STORM symposium, which aims to bring together expertise from fields of biology and physics as well as microscope software engineers to discuss the challenge of Stochastic Optical Reconstruction Microscopy (STORM).
This symposium is aimed at those researchers who are currently using STORM. We aim to provide a platform for discussion and collaboration to highlight the problems researchers are facing with STORM; and together we can try to find solutions.
There will be talks from researchers, addressing issues such as sample preparation, labelling and image analysis. As well as talks from experts including Nikon, and those using the Thunderstorm imageJ plugin. We encourage you to bring along a A3/A2 poster of a current image. These images do not have to be perfect, please bring along images you are having problems with or have questions about. See website for details. The images will be displayed during the day to allow you to get feedback from other attendees and prizes will be awarded to the most interesting. Registration closes 8th January 2017 but places are limited.
Registration is now open – please check our website for details:
Cytokinetic Abscission In the final step of cell division, the bridge connecting the cells is cut to give rise to two separate daughter cells – a fascinating process I have been working on since I started my PhD. This is a variation of my very first science-themed drawing, which I overlaid with an immunofluorescence staining labeling microtubules (green) and DNA (blue) – a combination of a hand-drawn illustration with real microscopy images that quite literally fuses science and art.
I have always loved science, and have always loved art – I combine these passions to illustrate scientific themes with an artistic twist. With my illustrations, I aim to highlight fundamental scientific aspects in an unconventional and refreshing way. I want to add some creativity to the conventional forms of scientific communication, with the aim to spark interest inside and outside the scientific community. I create my drawings for everyone to enjoy – for scientists to appreciate biological findings in a less serious way, and for non-scientists to grasp fundamental biological principles.
I used to draw a lot before studying Molecular Biology at the University of Vienna and the ETH Zürich in Switzerland. I recently completed my PhD in Daniel Gerlich’s group at the Institute of Molecular Biotechnology (IMBA), during which I made my very first science-themed drawing back in 2013. My PhD research focused on cytokinetic abscission, the final step of cell division (shown above, click on images for full size). When I showed that drawing as part of my scientific presentations, I realized that it sparked interest and tended to stay in people’s memories. This is when I discovered that adding an artistic twist to science creates a unique way to communicate science.
An Unconventional Take on Scientific Presentations. This picture was taken during my presentation for the Kirsten Peter Rabitsch Award, which I had the honor to receive for my PhD research earlier this year – in it you can see a new version of my very first science drawing.
An Unconventional Take on Scientific Presentations.This picture was taken during my presentation for the Kirsten Peter Rabitsch Award, which I had the honor to receive for my PhD research earlier this year – in it you can see a new version of my very first science drawing.
Being part of an institute that encourages creativity has helped me immensely to develop my artistic approaches. I participated in yearly campus-wide ‘Art & Science’ contests, where I experimented with drawing portraits of my colleagues and making a dress to illustrate my research project.
When Devotion Begets Emotion. These portraits of my fellow PhD students illustrate the intense emotions researchers face in everyday life in the lab. These drawings were part of a contribution to the Art & Science contest at the Vienna Biocenter, for which my team received the first prize in 2013.
The ‘ESCRT’ Dress. Cytokinetic abscission is mediated by a machinery composed of the Endosomal Sorting Complex Required for Transport (ESCRT)-III, which forms polymers that constrict the intercellular bridge until the membranes split. I created this “ESCRT” dress to illustrate how ESCRT-III separates the emerging daughter cells during abscission.
The ‘ESCRT’ Dress. Cytokinetic abscission is mediated by a machinery composed of the Endosomal Sorting Complex Required for Transport (ESCRT)-III, which forms polymers that constrict the intercellular bridge until the membranes split. I created this “ESCRT” dress to illustrate how ESCRT-III separates the emerging daughter cells during abscission.
I began pursuing scientific art after having my drawing selected for the cover and abstract book of the Cell Cycle Meeting in Cold Spring Harbor. The positive feedback I received there was an incredibly rewarding experience that encouraged me to start creating artwork for other people’s research as well as my own. Since then, I continue making scientific illustrations, one of which was recently featured on the EMBO Journal cover.
This EMBO Journal cover accompanies a paper on mammalian brain development I was involved in during my Master’s Thesis. The compass represents how the angle of the mitotic spindle in dividing cells affects their ultimate position within the brain – similar to a compass guiding the way to a location on a map.
EMBO Journal Cover.This EMBO Journal cover accompanies a paper on mammalian brain development I was involved in during my Master’s Thesis. The compass represents how the angle of the mitotic spindle in dividing cells affects their ultimate position within the brain – similar to a compass guiding the way to a location on a map.
I also began making artistic interpretations of recent scientific discoveries. Below is a small gallery of my illustrations for press releases that highlight recent publications.
Before chromosomes can be segregated during cell division, they have to cover themselves with a protein that acts similar to soap to prevent them from sticking together. This drawing is part of a press release for a recent discovery of a protein that acts as a surfactant to disperse chromosomes during anaphase.
The ribosome is a fascinating molecular machine that produces proteins. It reads the genetic code on our messenger RNA and translates them into proteins by linking the correct amino acids into a long chain. The ribosome consists of two huge subunits that twist against each other to drive this process of translation
Aside from highlighting research findings, I also began making illustrations for other purposes. For a popular science magazine, I created a less serious drawing illustrating the myth of the “five-second rule”, which suggests that bacteria will wait patiently before contaminating food that has been dropped on the ground.
Illustration for an article in the German version of Scientific American ‘Spektrum der Wissenschaft’.
I also had the wonderful opportunity to design the poster for the PhD symposium at the Vienna Biocenter titled ‘Mind the App’, as part of the organizing committee. For this illustration, as well as many of my others, I started by making a hand-drawn black and white sketch using pencils, and overlaid the colors digitally afterwards. I then added the apps on the phone to highlight the diverse applications of basic research that were covered at the conference.
Basic research gives rise to many ‘applications’. Poster of the ‘Mind the App’ VBC PhD Symposium at the Vienna Biocenter.
Basic research gives rise to many ‘applications’. Poster of the ‘Mind the App’ VBC PhD Symposium at the Vienna Biocenter.
When I first started drawing, I mainly focused on creating portraits – in this illustration I got back to my roots to make a short animation about everyday life in the lab.
Failed Experiment. An unsuccessful experiment can bring up very intense emotions, which every scientist is certainly familiar with. I created these drawings for an animation to be used in a video for the PhD Program at the Vienna Biocenter
I love the challenge of capturing the essence of scientific discoveries in an aesthetic and abstract way, and I am very excited for many more artistic adventures to come. Every drawing is an experiment!
On April 3-4, 2017 scientists from around Europe will be converging on Munich, Germany for the next meeting in the Abcam Brain meeting series – Programming and Reprogramming the Brain. Organizers, Benedikt Berninger (Johannes Gutenberg University Mainz) and Paola Arlotta (Harvard University) have put together a fantastic line up of speakers (see preliminary program).
This two-day meeting will provide a forum for presentations and discussions on the emerging field of brain development, reprogramming and modeling with a focus on new genome wide tools to understand biological processes with single-cell resolution.
Call for abstracts
Talk and poster places are available, so don’t wait, submit your abstract today!
Talk deadline: December 15, 2016
Poster deadline: February 6, 2017
We hope to see you in beautiful Munich next year!
Meeting topics
Modeling human brain development from pluripotent stem cells
Programming and maintenance of cell identity in the CNS
Development-inspired reprogramming of the brain
Decoding CNS complexity with single-cell resolution
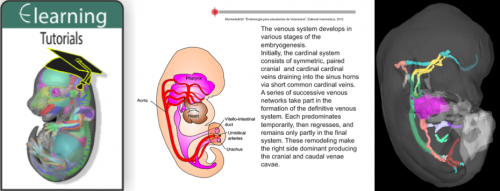
A new eLearning resource that provides short and interactive vignettes in embryo (primarily vertebrate) development, from gametogenesis through to organogenesis, is available from the eMouseAtlas1,2 website (www.emouseatlas.org).
The current eLearning content are the tutorials produced by Professor José García Monterde of the University of Córdoba, and the presentation from collaboration between Professor Monterde and the eMouseAtlas project.
The resource describes the development of various anatomical systems including the cardiovascular system, nervous system, and musculo-skeletal system, and additionally introduces core developmental biology concepts such as gastrulation, placentation, and the formation of the germ layers.
eLearning combines animations that enable conceptual understanding of key principles of embryonic development with 3D visualisations of embryo anatomy.
The resource combines illustrative animations of embryonic development with interactive 3D visualisations of mouse developmental anatomy. The newly developed 3D viewer enables arbitrary sections to be sliced through embryo models.
The eLearning tutorials additionally link to gene expression patterns associated with developing organ systems, the cellular-resolution eHistology atlas and the DMDD (DMDD.org.uk) database of embryonic lethal phenotypes.
eLearning is freely available for both teaching and research purposes. eMouseAtlas continues to develop tools and resources that enable students and researchers to understand the concepts underpinning embryonic development. In this context we are willing to host learning materials on behalf of the community.
References
Armit C, Richardson L, Hill B, Yang Y, Baldock RA (2015) eMouseAtlas Informatics: Embryo Atlas and Gene Expression Database. Mamm Genome. 26:431-40. doi: 10.1007/s00335-015-9596-5
Richardson L, Venkataraman S, Stevenson P, Yang Y, Moss J, Graham L, Burton N, Hill B, Rao J, Baldock RA, Armit C (2014).EMAGE mouse embryo spatial gene expression database: (2014 update) Nucleic Acids Res. 42(1):D835-44. doi: 10.1093/nar/gkt1155
Job Location: Brigham and Women’s Hospital, Development of Musculo-Skeletal Axis/Pourquié Lab, Boston MA
JOB SUMMARY:
Working under the general supervision of the Principal Investigator and on a day-to-day basis instructed by the Lab manager and Project supervisor(s) to contribute to research activities and lab operations to reach scientific goals. In accordance with established hospital policies and procedures, executes experiments involving in vitro protocols and cellular models for research studies targeting skeletal muscle, spine, dermis (paraxial mesoderm) development. Work additionally with in vivo embryo experimental systems (mouse, chicken). In addition, responsible for maintaining a moderately complex murine colony, laboratory supply inventory, cell culture room operation, and supporting lab safety compliance.
Principal duties and responsibilities will be:
Conducts research protocols autonomously and/or in team involving in vitro protocols and cellular models for research studies targeting skeletal muscles, spine and dermis (paraxial mesoderm) development.
Using aseptic technique, maintains tissue cultures and manipulates cell lines in a variety of complex experimental conditions including live-cell microscopy/imaging, library screens, and flow cytometry.
Tasks include processing cell culture, embryonic tissue for live-cell or histologic examination and conducting subsequent immunohistochemical and molecular analyses. Perform procedures such as DNA extraction, conventional PCR and real time PCR, western blotting, and time-lapse imaging.
Establishes and maintains a moderately complex mouse colony for the laboratory with responsibility for proper care, counts, inventory. May performs injections, some surgical procedures, microdissection and necropsy, following established and approved protocols. In collaboration with Project supervisor(s) , establishes new and modifies existing research methodologies and protocols.
Collect and Analyzes data , assists with data preparation toward manuscript publication and grant applications.
Documents accurately experimental work and research output
Reports and discusses progress of work regularly with Project supervisor and Principal Investigator.
Maintains lab supplies and reagent inventories
Coordinates lab compliance with institutional policies and procedures in the areas of safety, environmental and infection control, under Lab manager supervision
Assist in orientating and training new lab members, rotation students in area of expertise when required.
Perform all other duties and responsibilities as directed, including both research and administrative duties.
Qualifications:
Master of Science in a biological science required.
Minimum 3 year hands-on experience with cell culture assays, preferably pluripotent stem cells.
Experience with laboratory mouse colony maintenance helpful, but not required.
Skills/Abilities/Competencies Required:
Sound analytical and organizational skills.
High degree of computer literacy.
Careful attention to detail with good, detail-oriented observational skills.
Willingness and ability to conduct in vivo (mouse) experiments.
Excellent oral and written communication skills.
Must have sound interpersonal skills. Ability to constructively interact with members of a research team to pursue scientific goals is a necessity.
Ability to work closely with Project supervisor (s)
Working Conditions:
Normal research laboratory environment.
Exposure to laboratory reagents, chemicals, and animal and human tissues under controlled conditions. Minimal risk when following established protocols and federal, state, and hospital guidelines.
To apply to this position please send your resume and cover letter to jchal@partners.org
The Company of Biologists is a not-for-profit publishing organisation dedicated to supporting and inspiring the biological community. We currently have two opportunities available in our Cambridge office.
Journal Website Content Manager
We are creating an exciting new role to enhance the community content on our journal websites.
We publish five important journals that serve the biological research community. All have effective publishing platforms and a good social media presence. We now seek to extend our community engagement, raise awareness of our charitable activities and build connections with early career scientists.
DMM is looking for an enthusiastic intern who wishes to gain experience in science publishing, including writing press releases, contributing to our social media activities, and supporting our Reviews Editor with commissioned articles. The internship is envisaged to last for 9 months at a salary of £20,000 per annum pro rata.
Our interns have a great track record of continuing on into important publishing roles.
Drosophila hematopoiesis shows striking resemblance with that of vertebrates, both at the level of signalling molecules and the phase of development. Even though there has been no report of Hematopoietic stem cells (HSCs) in Drosophila, this model has been employed extensively to understand progenitor biology and niche interactions. The Drosophila blood cells are specialised precursor cells (present within the hematopoietic organ: the lymph gland), that show several similarities with stem cells. They are known to differentiate into mature blood cells of the fruit fly. Our new study identifies the “founder cells” or the “stem cells” that give rise to these specialised blood-progenitors.
How it started off:
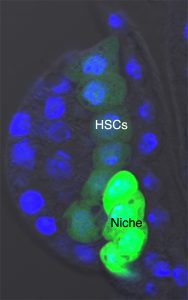
Our team was involved in detailed characterisation of signals which are required for the maintenance of the larval hematopoietic niche. To understand the dynamicity of the expression pattern of a battery of niche markers, we ventured into dissecting the larval lymph gland of the early first instar. Analysing a newly emerged larvae, about 8 hrs old, we encountered a group of large cells that always aligned themselves near the dorsal vessel or larval heart. We started calling them “Big Cells”. It turned out that these cells expressed several markers seen in other described stem cells.
Founder Cells or HSCs:
The very fact that they stood out almost every time in a early instar lymph gland caught our attention, and we felt tempted to characterise them. Our attempt to trace them throughout development revealed that although the “Big Cells” do not die, they could not be tracked beyond 22 hours post hatching. We argued that maybe they were changing their fate. Indeed, on lineage tracing of these cells and analysing them at various time point of development, we could see that they are actually converted to blood progenitor cells. Analysing the same genotype at mature third instar larval stages indicated that not only progenitors but also differentiated cells are lineage traced. This clearly indicates that the “Big Cells” are multi-potent in their nature. As a functional correlate, we genetically ablated the “Big Cells” and analysed the consequence on the mature lymph gland. Remarkably, the elimination of these 4-5 “Big Cells” drastically reduced the size of the gland.
The first instar larval lymph gland with the HSCs and its Niche marked by Dorothy-GFP expression
The final climax:
At this point we were thrilled that we had a fine story to tell about the elusive founder cells of Drosophila lymph gland. But a stem cell story without niche is not complete! We therefore entered into a spree to nail down the niche and the signal. Our suspicion was on the already described progenitor niche known to maintain the precursor cells. Since this niche is specified in embryo and is the most differentiated cell type in the first instar lymph gland, we speculated that it might also be serving as the niche for the HSCs. Our expression studies demonstrated that HSCs are enriched in phosphorylated Mad (pMAD), a pathway effector of Dpp signalling. To start with, therefore, we down regulated pMad from the HSCs. This resulted in their disappearance and a concomitant reduction in lymph gland size. Strikingly, on knock down of Dpp signalling from the progenitor niche, the HSCs failed to self-renew, clearly validating that Dpp signalling from the progenitor niche is its source of sustenance. It is to be noted that vertebrate early HSCs are also maintained by BMP (vertebrate counterpart of Dpp) signalling.
What are the implications:
With the identification of the early HSCs and the signal that maintains in Drosophila larvae, we have not only demonstrated the conservation with vertebrates, but put forward a model that can be used for answering several questions pertinent to vertebrate early blood development, both in normal as well as aberrant conditions.
Drevon, C., and T. Jaffredo. 2014. “Cell interactions and cell signaling during hematopoietic development.” Exp Cell Res 329 (2):200-6. doi: 10.1016/j.yexcr.2014.10.009.
Durand, C., C. Robin, K. Bollerot, M. H. Baron, K. Ottersbach, and E. Dzierzak. 2007. “Embryonic stromal clones reveal developmental regulators of definitive hematopoietic stem cells.” Proc Natl Acad Sci U S A 104 (52):20838-43. doi: 10.1073/pnas.0706923105.
Evans, C., Olson, J., Ngo, K., Kim, E., Lee, N., Kuoy, E., Patananan, A., Sitz, D., Tran, P., Do, M., Yackle, K., Cespedes, A., Hartenstein, V., Call, G., & Banerjee, U. (2009). G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila Nature Methods, 6 (8), 603-605 DOI:10.1038/nmeth.1356
Evans, C.J., Sinenko, S.A., Mandal, L., Martinez-Agosto, J.A., Hartenstein, V., & Banerjee, U. (2007). Genetic Dissection of Hematopoiesis Using Drosophila as a Model SystemAdvances in Developmental Biology, 18, 259-299 DOI: 10.1016/S1574-3349(07)18011-X
Jung, S. H., C. J. Evans, C. Uemura, and U. Banerjee. 2005. “The Drosophila lymph gland as a developmental model of hematopoiesis.” Development 132 (11):2521-33. doi: 10.1242/dev.01837.
Mandal, L., Banerjee, U., & Hartenstein, V. (2004). Evidence for a fruit fly hemangioblast and similarities between lymph-gland hematopoiesis in fruit fly and mammal aorta-gonadal-mesonephros mesoderm Nature Genetics, 36 (9), 1019-1023 DOI: 10.1038/ng1404
Wellcome exists to improve health for everyone by helping great ideas to thrive. We’re a global charitable foundation, both politically and financially independent. We support scientists and researchers, take on big problems, fuel imaginations and spark debate.
We are seeking up to two Science Portfolio Advisers or Developers to contribute to managing the Cellular and Developmental Sciences portfolio within our Science division. You will liaise with internal and external stakeholders, including other funders, and provide our community of researchers with support and advice. You will need to keep abreast of the scientific field and attend key national and international conferences in the field.
To be successful in this role you will need to have a PhD and postdoctoral scientific experience in a relevant discipline. Some experience working outside academia in a research, funding or policy role is desirable although not necessary.
You should be confident interacting with your peers and the external scientific community. You will also be able to demonstrate that you can:
see the big picture and recognise scientific potential and opportunities
assimilate complex issues and work across science areas
lead and manage teams
communicate effectively and confidently with individuals and groups
work effectively and cooperatively within a team/matrix structure
apply your strong analytical and written skills to the development of briefing/position documents.
The salary will be between £36,000 and £52,000 plus benefits, depending on your experience.
To apply, please complete the online application process and ensure your covering letter explains how you meet the criteria for this job.
 (No Ratings Yet)
(No Ratings Yet)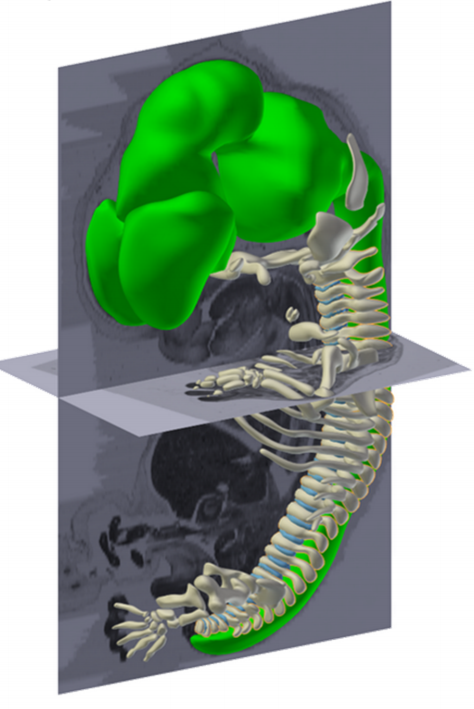
 (2 votes)
(2 votes)